
Chapter 1
Principles of Pharmacology and Toxicology
The right dose differentiates a poison and a remedy.
Paracelsus, 1493–1541
Introduction
The past century has seen a tremendous expansion in the number of synthetic chemicals employed by humankind as materials, drugs, preservatives for foods and other products, pesticides, cleaning agents, and even weapons of war. The American Chemical Society maintains a chemical registry. Since 1907, it has recorded 33 million organic and inorganic substances and 58 million sequences as of 2008. About 4000 new chemicals are added each day. A study by the Danish Environmental Protection Agency calculated that 13.4% of them possess acute toxicity, 2.5% reproductive toxicity, 3.9% are mutagens, 1.8% carcinogens, and 3.5% are dangerous to the aquatic environment. Four thousand chemicals are used as medicinals and at least 1200 more as household products. Add to this the numerous natural substances, both inorganic and organic, that possess toxic potential, and it is little wonder that the public expresses concern and even, sometimes, panic about the harmful effects these agents may exert on their health and on the environment. Tens of thousands of these agents have never been subjected to a thorough toxicity testing.
According to the Danish study, thousands of chemicals are potential carcinogens but the number that has been confirmed to be human carcinogens is much smaller. About 500 chemicals have been evaluated for carcinogenic potential. Some 44 have been designated as possible human carcinogens on the basis of evidence, either limited or conclusive, obtained from human studies. Of these, 37 were tested positive for carcinogenicity in animal tests and were later shown to be carcinogens for humans. There are, however, numerous other agents that have been shown to be carcinogenic in rodents but that are yet to be identified as human carcinogens. This creates significant problems regarding the legislative and regulatory decisions that need to be made about their use. Some of the areas of uncertainty that surround the extrapolation of data from the animal setting to the human one will be discussed in the following chapter. The process of extrapolation requires input from many different disciplines that may include engineering, physics, biology, chemistry, pathology, pharmacology, physiology, public health, immunology, epidemiology, biostatistics, and occupational health. The field of toxicology thus depends on all of these, but perhaps draws most heavily on pharmacology, biochemistry, and pathology. It is the identification of the degree of risk to which individuals or groups are exposed in a given set of circumstances that directs all of this activity.
Other forms of toxicity, hepatotoxicity, nephrotoxicity, and neural toxicity, for example, may be more important in acute exposures that might occur in the industrial setting. Reproductive and fetal toxicity has been demonstrated frequently experimentally, but their significance for the general population exposed to low levels of toxicants in the environment remains unclear.
The (U.S.) Agency for Toxic Substances and Disease Registry, and The (U.S.) Environmental Protection Agency jointly maintain a priority list of toxic substances. The “top 15” in the 2011 list are arsenic, lead, metallic mercury, vinyl chloride, polychlorinated biphenyls (PCBs), benzene, cadmium, benzo[a]pyrene, polycyclic aromatic hydrocarbons (PAHs), benzo[b]fluoranthene, chloroform, aroclor 1260, P′P′-DDT, aroclor 1254, and dibenz[a,h]anthracene. The complete list can be viewed on the Internet at www.atsdr.ctv.gov “Priority List of Hazardous Substances.”
Considerable difficulty attends efforts to extrapolate the results of toxicity tests in experimental animals to humans exposed to very low levels in their environment, especially with regard to the risk of cancer. Current legislation requires testing in two species with sufficient numbers for reliable statistical analysis. Rats and mice are generally used, as hamsters are resistant to many carcinogens and primates are too expensive and, in the case of some species, too environmentally threatened. For statistical purposes, cancer includes all tumors whether benign or malignant. A 2 year carcinogen study employing two species cost, in 2010, at least $2,000,000 plus the costs of 1 year for preparation, one for analysis (pathology, etc.), and one for documentation and statistics. Since it is not practical to test every chemical, several factors need to be considered in selecting test chemicals. These include the frequency and severity of observed effects, the extent to which the chemical is used, its persistence in the environment (examples of persistent chemicals include chlorinated hydrocarbons), and whether transformations to more toxic agents occur.
Heavy metals, the by-products of most mining and ore extraction processes, are examples of ubiquitous toxicants with almost infinite half-lives. Mercury (Hg), for example, is present in all canned tuna at about 5 parts/million (ppm), mostly from natural sources. Aquatic bacteria can transform mercury to methylmercury. This has a different toxicity profile. Cadmium (Cd) enters the environment at about 7000 ton/year and is concentrated by livestock because they recycle it in feces used for fertilizer. It is then passed on to forage grasses. Radioactive isotopes of cesium and iodine entered the food chain after Chernobyl.
The estimation of the degree of risk associated with the presence of a potentially toxic substance in the environment is the basis for all decisions relating to the legislative controls over that chemical, including its industrial use and eventual disposal. Pharmacological/toxicological principles are essential for understanding the processes involved in toxicity testing.
Pharmacology may be defined as the science of drugs. It includes a study of their sources (materia medica); their actions in the living animal organism (pharmacodynamics); the manner in which they are absorbed, moved around in the body, and excreted (pharmacokinetics); their use in medicine (therapeutics); and their harmful effects (toxicology). In this context, a drug is any substance used as a medicine but pharmacology generally includes the study of substances of abuse and, in the broadest sense, deals with the interactions of xenobiotics (literally, substances foreign to living organisms) whether they be natural or man-made (anthropogenic), therapeutic or toxic. In this sense, toxicology can be considered to be a branch of pharmacology. Xenobiotics may also be exploited as research tools to reveal mechanisms underlying physiological processes.
Toxicology is the study of the harmful effects of xenobiotics on living organisms, the mechanisms underlying those effects, and the conditions under which they are likely to occur.
Environmental toxicology is the study of the effects of incidental or accidental exposure of organisms, including human beings (the focus of this text), to toxins in the environment, i.e., air, water, and food. While the greatest concern today centers on pollutants of human origin, it should not be forgotten that toxic substances, including carcinogens, abound in nature. The subject of environmental toxicology embraces the study of the causes, conditions, environmental impact, and means of controlling pollutants in the environment. It may also be extended to include the environment of the workplace (industrial hygiene). The related term ecotoxicology deals with the harmful effects of chemicals, usually of anthropogenic origin, on ecosystems.
Economic toxicology is the study of chemicals that are developed expressly for the purpose of improving economic gain by selectively eliminating a species (insecticides and herbicides), improving health and productivity (drugs), preserving foodstuffs (food additives), or for the manufacture of a marketable product (industrial solvents, cleaning agents, etc.).
Forensic toxicology refers to the medico-legal aspects of the harmful effects of drugs and poisons administered or taken deliberately or accidentally. Detection of xenobiotics in tissues and fluids and in, or on, objects is an important aspect of this field as is the preparation of evidence for submission in court.
Pharmacokinetics
There has been a trend recently to attempt to separate toxicology from pharmacology by the use of such terms as toxicokinetics, toxicodynamics, etc. The distinction is largely semantic. The principles governing the absorption, distribution, metabolism, and excretion of a xenobiotic are the same regardless of whether it is used as a therapeutic agent or is a toxin. Throughout this text, pharmacological can be taken also to represent toxicological.
The response of organisms to drugs and chemicals is governed by natural laws. One of these is the Law of Mass Action, which dictates that, in the absence of a transport system, chemicals in solution will move from an area of high concentration to one of low concentration. If a semipermeable membrane is interposed between these areas, the chemical will move across it, assuming the chemical can penetrate the membrane. In reality, molecules wander randomly across the barrier, but the frequency of transfers will be greater from the area of high concentration to that of the low one until equilibrium is established. Cell walls and other biological membranes function as semipermeable membranes, and The Law of Mass Action influences the uptake of most drugs and toxicants by living organisms. The concentration of a toxicant in the environment (water, air, soil) is thus an important determinant affecting its uptake. Transport mechanisms are dealt with under “Absorption” and “Distribution” sections.
Partition coefficient is the ratio of a chemical’s relative solubility in two different phases. The ratio of solubility in oil (often n-octanol) to that in water is frequently used to predict the distribution of a xenobiotic between the aqueous and lipid phases in the body.
Absorption
Whether or not a xenobiotic is toxic, and how that toxicity is manifested, depends largely on how the body deals with it. Substances that are not absorbed from the gastrointestinal tract have no systemic toxicity. This fact allows barium to be used as an x-ray contrast medium, despite barium’s toxicity by other routes of administration. The selective toxicity of most insecticides depends solely on a greater ability to penetrate the chitin of the insect’s exoskeleton than to penetrate human skin. A substance that is not readily excreted by the body (usually through the kidneys or in the feces) will accumulate to toxic levels.
The primary routes of absorption for toxicants are the skin, the lungs, and the gastrointestinal tract. The latter two are important for the population at large but the skin may be a very significant site in certain industrial settings. The site of absorption, more commonly called the portal of entry in toxicology, can have a significant influence on the toxicity of a substance.
Larger molecules require a degree of lipid solubility to cross biological barriers since cell membranes consist of a fluid phospholipid matrix with embedded proteins that may penetrate partway or all the way through the membrane. Factors that influence the lipophilicity of a chemical will therefore affect its absorption. Many chemicals are weak acids or bases that may exist in an ionized (polar) or a nonionized (nonpolar) state with equilibrium established between them; for example,
R−HNonpolar→R−+H+Polar
The polar form is water soluble whereas the nonpolar form is lipid soluble. The pH will influence the equilibrium and, hence, the amount of the lipid-soluble form that is available for absorption. The dissociation constant (pKa) of a substance is defined as that pH at which 50% of it will exist in each state. Weakly acidic drugs are shifted to the nonpolar state in an acid medium and to the polar state in an alkaline medium. The reverse is true for weakly alkaline drugs. The pH of the stomach and upper small bowel is acidic (pH 2–4); therefore, acidic chemicals will be absorbed here. Alkaline substances tend to be absorbed in the lower small bowel and the upper colon that are more alkaline, whereas the descending colon becomes acidic again.
Lipid solubility is not essential for the passage of all molecules across membranes. There is the bulk transfer of water across the cell membrane that can carry very small (less than 200 Da) water-soluble molecules with it. Metallic ions such as calcium, sodium, and potassium, as well as chlorine, can pass through special channels, some of which are regulated by the transmembrane potential (voltage-regulated) and others by specific receptors (receptor-activated). Specialized exchangers also exist, for example, the sodium pump.
Active transport is an energy-consuming process by which a substance may be moved against a concentration gradient. Active transport is important in the kidney and the liver. In addition to energy consumption, it is also characterized by saturability, selectivity for specific chemical configurations, and the ability to move substances against an electrochemical gradient. Facilitated diffusion is similar except that no energy is consumed, and it cannot occur against an electrochemical gradient.
Pinocytosis is a process whereby a segment of the plasma membrane of a cell invaginates to form a sack in which extracellular fluid and colloidal particles can be taken into the cell by pinching off the “mouth” of the sack. This is an important mechanism by which the mucosal cells of the intestinal tract take up nutrients and some drugs and chemicals.
Distribution
Once absorbed, the agent may be distributed throughout various compartments in the body. Serum albumin possesses many nonspecific binding sites for xenobiotics, especially weakly acidic ones, and it therefore becomes a transport system for many substances. The balance between dissociated (polar) and undissociated (nonpolar) states also affects the distribution of a chemical as well, since pH changes from the extracellular fluid (pH 7) to the plasma (pH 7.4). The partition coefficient of a substance also influences its distribution, determining, for example, the extent to which it will be sequestered in fat. Highly lipid-soluble substances will be sequestered in body fat, where they may remain for long periods. Everyone has DDT and its metabolites dissolved in their fat. The amount varies with their age and location.
The use of DDT in North America was drastically reduced in the 1970s and a complete ban was legislated in Canada in 1990. Substances like DDT, which are sequestered in fat, may be released during periods of starvation, extreme dieting, as a result of illness, and even during lactation, when lipids are transferred to milk. The released toxicant may reach concentrations at target sites sufficient to cause a toxic response. Figure 1.1 illustrates these relationships among storage fat, blood, and target organ.

Disposition of lipid-soluble chemicals in adipose tissue and blood and the effect of severe weight loss.
The rate of distribution of a substance is a function of the rate of blood flow through the tissues (tissue perfusion). Highly vascular organs will accumulate it first; organs that are poorly perfused will accumulate it last. The substance is thus distributed initially on the basis of tissue perfusion, then, as equilibrium states are reached, it will redistribute on the basis of its solubility. Following the intravenous injection of a chemical with a high partition coefficient, equilibrium will be established instantly with the kidney and liver because of their high vascularity, almost as quickly with the brain, with muscle in about 30 min, and with fat in about 3 h. The membranes surrounding the brain and separating it from its blood vessels constitute the blood–brain barrier that generally will pass only quite lipid-soluble agents such as all anesthetics.
Thus, tissue perfusion and partition coefficient may play important roles in determining the onset and termination of either a therapeutic or a toxic response. Sodium thiopental, an ultrashort-acting barbiturate, is used for anesthetic induction. The rate of biotransformation is so slow as to have little effect on recovery. The drug readily penetrates the blood–brain barrier because of its high lipid solubility and the brain, which is richly perfused, rapidly takes it up and anesthesia ensues. This effect is terminated because the drug is redistributed to other tissues, including depot fat, which is poorly perfused. New equilibria are established among blood, brain, and other tissues so that, while initial recovery is rapid, a state of sedation may persist for several hours. In Figure 1.2, the effects of perfusion and partition coefficient on T1/2 s of thiopental in different tissues are shown.

Tissue T1/2 values for sodium thiopental, a highly lipid-soluble, ultrashort-acting barbiturate anesthetic.
Biotransformation
Biotransformations of xenobiotics are classified as either Phase I reactions or Phase II reactions.
Phase I chemical reactions, also known as nonsynthetic biotransformations, convert a lipophilic (fat soluble) substance to a more polar, and hence more water-soluble, substance. This metabolite is excreted more readily by the kidneys than the parent compound, but it usually retains significant bioactivity. It may be more active, or less active, than the parent substance. If the parent chemical is nontoxic but the metabolite is toxic, this is a toxication reaction. A drug that requires biotransformation to become active is referred to as pro-drug. Figure 1.3 shows some examples of Phase I reactions and their consequences.

Some examples of Phase I reactions. The product may be more active or less active than the parent compound or it may be inactive.
Phase I chemical reactions include oxidation, reduction, and hydrolysis and generally unmask or introduce a functional (reactive) group such as –NH2, –OH, –SH, COOH. The oxidation reactions are listed in the next paragraph dealing with the cytochrome P-450 enzyme group. Hydrolysis of esters and amides also occurs. Reduction reactions may involve azo (RN = NR) or nitro (RNO2) groups.
Many oxidation reactions are under the control of a group of mixed-function oxidases for which members of the cytochrome P450 (CYP450) group serve as a catalyst. CYP450 enzymes are widely distributed in nature and are involved in the biotransformation of a multitude of xenobiotics as well as numerous naturally occurring substances. They are located primarily in the smooth endoplasmic reticulum (SER) of hepatic cells, but they exist in many tissues as well as many species, including single-celled organisms. The CYP450 monooxygenases have tremendous substrate versatility, being able to oxidize lipophilic xenobiotics plus fatty acids, fat-soluble vitamins, and various hormones. This is partly because there are at least 20 variants of the enzyme (isozymes) and because each is capable of accepting many substrates. CYPs 1, 2, and 3 are isozymes especially involved in xenobiotic transformations. The reactions they catalyze include aromatic and side-chain hydroxylation; N-, O-, and S-dealkylation; N-oxidation; N-hydroxylation; sulfoxidation; deamination; dehalogenation; and desulfuration. There are likely 1000 or more CYP450 isozymes in nature with perhaps 50 that have significance in mammals.
It should be noted that pro-carcinogens are converted to carcinogens by Phase I reactions. Examples of this include benzo[a]pyrene, the fungal toxin aflatoxin B1, and the synthetic estrogen diethylstilbestrol. This process often involves the formation of an epoxide compound, as it does in the three examples given. An epoxide has the chemical configuration shown in Figure 1.4, making it highly nucleophilic and chemically reactive. Many epoxides are carcinogens. Figure 1.4 shows this chemical transformation for stilbestrol and benzo[a]pyrene, which is an example of a polyaromatic hydrocarbon (PAH). Many of these are carcinogenic and are environmental pollutants. Other enzymes called epoxide hydrolases may detoxify the epoxides.

Phase II reactions are conjugation (synthetic) reactions that render the agent not only more water soluble, but biologically inactive, with a very few exceptions. A common conjugation reaction is with glucuronic acid. Conjugation also occurs with sulfuric acid, acetic acid, glycine, and glutathione. Many Phase I metabolites are still too lipophilic (fat soluble) to be excreted by the kidneys and are subjected to Phase II conjugation. All chemicals need not be subjected first to Phase I transformations. Many, if they possess the necessary functional groups (e.g., –OH, –NH2) are conjugated directly.
An important concept for understanding toxication and detoxication of xenobiotics is enzyme induction. Hepatic enzymes of the SER can be stimulated to a higher level of activity by many highly lipophilic agents. Because these enzymes are nonspecific, this has consequences for many other agents transformed by the same enzymes. Induction is accomplished by the increased synthesis of more enzymes, so the SER actually increases in density. The result may be increased detoxication of a chemical, or the increased synthesis of a toxic metabolite. Cigarette smoke contains many inducers and may increase the breakdown of many drugs (theophylline, phenacetin, etc.) but conversely it may act through this mechanism as a promoter or as a co-carcinogen.
Elimination
Every secretory or excretory site in the body is potentially a route of elimination for xenobiotics. Thus, they may be excreted in saliva, sweat, milk, tears, bile, mucus, feces, and urine. Of these, the most significant site is urine, followed by feces and bile. The kidney (Figure 1.5) is the principal organ for the elimination of natural waste metabolites. Most of these are toxic if they exceed normal levels. The kidney also is the main organ for maintaining fluid and electrolyte balance. It is therefore not surprising that the kidney also is the main site of elimination of xenobiotics, including drugs. Although it constitutes only 0.4% of total body weight, it takes 24% of the cardiac output. It is a highly efficient filter of blood.

The basic physiological unit of the kidney is the nephron (see Figure 1.5), which is composed of the glomerulus (a tightly wound bundle of blood vessels) and the tubule, which is closed at the glomerular end to provide a semipermeable membrane. The tubule is composed of several segments with different functions. Molecules with a molecular weight less than 66,000 Da are passed through the glomerulus. They may be reabsorbed further down the tubule and even re-secreted. This occurs with uric acid, which is completely passed through the filter, 98% reabsorbed, and further secreted. The pH of urine will determine the degree of dissociation of acids and bases and hence influence their movement across the reabsorption sites. Manipulating urine pH is a method of accelerating the elimination of some xenobiotics. For example, increasing urinary pH from 6 to 8 can increase the excretion of salicylate by almost 1 order of magnitude.
Passive diffusion across the distal tubule depends on the degree of ionization in the plasma and extracellular fluid as only the lipid-soluble form will be diffused. The concentration gradient also thus is an important rate-limiting factor. Very-water-soluble agents are passed through the glomerulus if they are small enough, and this is the reason why most biotransformations result in increased water solubility. Other substances are actively secreted (an energy-consuming process) at tubular sites (see Figure 1.5).
It should be noted that the lungs are a very important site of elimination for volatile substances including solvents, alcohols, and volatile and gaseous anesthetics. These can, in fact, be smelled on the breath, which can be an important first aid procedure to determine the cause of unconsciousness or stupor. Ketoacidosis in diabetics also can be detected by the acetone-like, or fruity, odor on the breath. Young diabetics have been suspected of glue sniffing when brought to an emergency department in a stupor or coma because of this fact.
Many drugs and chemicals are excreted into the bile. These tend to be polar agents, both cationic and anionic, the latter including glucuronide conjugates. Nonselective active transport systems, similar to those in the kidneys, are involved in the excretory processes. Once they enter the small intestine, these chemical metabolites may be excreted in the feces or reabsorbed back into the bloodstream. Enzymatic hydrolysis of glucuronide conjugates favors a return to the more lipid-soluble state and hence reabsorption. Purgatives may sometimes be used to facilitate the elimination of chemicals from the bowels.
The excretion of xenobiotics in mother’s milk may not be an important route of elimination, but it can have significance for toxicity in the infant. The chloracne rash associated with the now-obsolete bromide sedatives appears to be related to the secretion of this halogen in sweat. It is distributed in the body like chloride ion.
Extensive batteries of enzymes in the body may render the chemical nontoxic (detoxication), more water soluble, and hence more easily excreted, or they may activate it to a toxic form (toxication). The liver is the primary site of xenobiotic biotransformation in the mammalian body, but it is by no means the only one. Indeed, significant biotransformation can occur at the portal of entry. The chemical pathways are often the same. The response of the body to chemical insult also depends on the mitotic activity of the target tissue. Rapidly dividing tissues allow little time for repair to occur before cell division, so that the chance of a mutation is increased. Moreover, tissues that regenerate poorly are vulnerable to permanent damage by toxicants.
Pharmacodynamics
Ligand Binding and Receptors
Since only the molecules that are free in solution contribute to the concentration gradient, their binding to tissue components, or their chemical alteration by tissue enzymes will contribute to the maintenance of the gradient. The nature and strength of the chemical bond determines how easily the xenobiotic will dissociate when the concentration gradient is reversed. Drugs interact with specific sites (receptors) on proteins such as plasma membrane proteins, cytosolic enzymes, membranes on cell organelles, and, in some cases, nucleic acids (e.g., certain antineoplastic drugs). Membrane receptors and enzymes have molecular configurations that will react only with certain molecules in a kind of “lock-and-key” manner. Ease of reversibility is an important characteristic for most drugs, so that as concentration of the free substance falls, the drug comes off the receptor and its effect is terminated. This is often expressed by the following equation:
Drug(D)+Receptor (R)←→DR complex←→Response
The magnitude of the response is determined by the number (percentage) of receptors occupied at any given time. Neither the drug nor the receptor is altered by the reaction, which is defined as pharmacodynamic. Drugs are generally classified as being agonists, partial agonists, or antagonists, and receptors can exist in an active or an inactive state. An agonist is a drug that promotes the active state because it has a high affinity for receptors in that state. A drug with modest affinity for the active state of the receptor will be a partial agonist and a drug with equal affinity for both the active and the inactive states will be a competitive antagonist; meaning that its antagonism can be overcome by a sufficient concentration of an agonist. An antagonist that binds so strongly to the active receptor that it cannot be reversed is a noncompetitive antagonist.
In many cases, drugs and toxicants interact with receptors that normally accept physiological ligands such as neurotransmitters, hormones, ions, and nutritional elements. The proteins of cell surface receptors may penetrate to the interior of the cell in the case of ion channels and exchangers, or they may connect with other proteins in the membrane to transduce signals. Many neurotransmitters operate through a family of receptors that share the property of connecting to a protein having seven, membrane-spanning peptide chains. These “G” proteins (G for guanosine triphosphate or GTP) are transducers that interact with enzymes such as adenlycyclase or phospholipase C to initiate intracellular second messengers. G proteins may be inhibitory (Gi), stimulatory (Gs), or operate through other, unidentified, mechanisms (Go). The neurotransmitters noradrenaline, acetylcholine, dopamine, serotonin, histamine, gamma-aminobutyric acid (GABA), glycine, and glutamic acid have been shown to act through G-protein receptors. Many centrally acting drugs work through these receptors.
Steroid receptors also exist. These are soluble cytosolic receptors that bind to the steroid after it diffuses into the cell and carry it to the nucleus. Opioid receptors in the CNS accept the endogenous peptide endorphins and encephalins. These receptors are the site of action of the narcotic analgesics.
Any receptor is a potential target for a toxicant interaction. A special case is the aryl hydrocarbon, or Ah, receptor. This cytosolic receptor binds to aromatic hydrocarbons like dioxins and it is believed that it is involved in their toxicity. No natural ligand for this receptor has yet been identified in mammals. This subject is discussed in detail in the chapter on halogenated hydrocarbons.
The chemical bond with the target receptor can involve covalent bonds, as well as noncovalent bonds including ionic, hydrogen, and van der Waal’s forces. If the xenobiotic interacts irreversibly with a component of a cell, the effect may be long-lasting. Indeed, irreversibility of effect is an important characteristic of many toxicants (organophosphorus insecticides are examples of irreversible inhibitors of the enzyme acetylcholinesterase). If a chemical reacts irreversibly with DNA, a mutation may result in carcinogenesis or teratogenesis. This effect is sometimes described as “hit-and-run” because it is unrelated to any measurable concentration of the agent in the serum (see the following text).
Irreversibility of binding does not always mean irreversibility of effect. The drug acetylsalicylic acid (aspirin) is an irreversible inhibitor of the enzyme cyclo-oxygenase, which accounts for many of its pharmacological actions. Provided that exposure to aspirin is terminated, the effect declines as new enzyme is synthesized.
Biological Variation and Data Manipulation
Within any given population of organisms, there will be some that will respond to a drug or toxicant at the lowest concentration, and others that only respond at the very highest concentration, whereas most subjects will be grouped around the mean response. This is true of all organisms, including human beings and single-celled ones. It is even true of populations of like cells (liver cells, kidney cells, blood cells) within the body, and may partly explain why some cells may become malignant while others do not. It is the existence of biological variation that necessitates the use of large populations of test subjects and the development of mathematical treatments of data to permit the comparison of different populations of test subjects. If the responses of the species in question are grouped symmetrically about the mean response, a “normal” or Gaussian distribution curve is obtained (Figure 1.6). In this case, 68.3% of the population will fall within ±1 standard deviation (SD) of the arithmetic mean, 95.5% between ±2 SDs, and 99.7% between ±3 SDs. A datum point lying outside these limits is assumed not to belong to the test population. Sometimes the population is skewed, however, with more subjects falling on one side of the mean than on the other. Factors accounting for variability could include differences in the rate and degree of uptake, distribution, biotransformation, and excretion and even the nature and number of binding sites and receptors for the agent. These factors may be under genetic control, or they could be due to environmental differences in such things as temperature, nutrition, disease, the presence of other xenobiotics including medications, and so on. They also tend to vary with age and sex.

Normal (Gaussian) distribution of a population responding to different drug doses. A theoretical normal or “Gaussian” dose response distribution curve for a drug.
Dose Response
A population distribution in response to a drug or chemical applies even to like cells within the same organism or cell culture, because some cells will be more aged than others or may be defective in some way. Therefore, it is impossible from a single dose of a drug or toxicant to draw any conclusions about its potency, since it is not known whether one is recruiting only the most sensitive cells, or nearly all of them. For this reason, it is necessary to construct a dose–response curve whether testing a new drug for its effective dose or a chemical for its toxicity. Typically, the response rises rapidly once the threshold is exceeded, and then flattens out as fewer and fewer cells remain to be recruited (Figure 1.6). This type of response is a graded or dose-dependent response. There is another type of response that can be described as yes/no or all-or-nothing, and this is a quantal response. Lethality is an example. For graded responses, it is important to establish standard points of comparison, since comparing a dose at the low end of the response for one chemical, with one at the top end for another is not statistically reliable. The point usually chosen is that dose which produces 50% of the maximum effect, the effective dose (or concentration) 50% (ED50 or EC50). “Dose” is used when the test substance is administered individually to the test subjects, and “concentration” when it is added to the surrounding medium, such as the water in an aquarium or the fluid bathing an isolated tissue in an organ bath.
A quantal response can be converted to a graded one by using several test groups, each receiving a different dose of the agent being tested. The percent of animals showing the expected response can then be plotted against dose. Thus, one can calculate the dose that, on average, will kill 50% of the test animals (lethal dose 50%, LD50). If the response is a toxic one (liver necrosis, kidney failure), the value is the toxic dose 50% (TD50). Variations on this approach include the LD10, TD10, etc. The LD1 is also called the Minimum Lethal Dose. Values such as the LD1 and the LD10 are replacing the LD50 in many jurisdictions.
In attempting to compare responses to two different chemicals, it is useful to perform a mathematical manipulation on the data so that differences or similarities in the shapes of the dose–response curves are more obvious. This involves plotting the logarithm of the dose against the response and this converts the exponential curve shown in Figure 1.7 to the sigmoidal one (S-shaped, Figure 1.8).


Semilogarithmic plot (response vs. the log of the dose) of the dose–response curve shown in Figure 1.7 .
It is now much easier to interpolate to the EC50 (since the selected doses might not have included it) and to compare these points. Using the log of the dose tends to overcome the fact that large increases in dose result in small increases in response on the right side of the curve, whereas small increases in dose result in large increases in response on the left side of the curve. Thus, the midpoint, the EC (or TD)50, provides the greatest statistical reliability. Parallel slopes of curves suggest similar mechanisms of action, and comparisons based on molar concentrations provide information on relative potencies. Toxicity comparisons may be done by calculating the Therapeutic Index (TI) if the substance is a therapeutic agent. This is the LD50/ED50; the higher the number, the safer the agent. Other estimates of safety, more appropriate to toxicity studies of nontherapeutic agents, involve comparisons of the TD1 and the EC99.
It is important to note that all toxicity tests contain a temporal factor in that the determination of toxic effects is conducted at a specific time after exposure. Acute toxicity studies generally involve determinations made 72 h after a single high dose, whereas long-term toxicity requires multiple exposures with measurements made at least 28 days later. These studies are defined by government regulations in jurisdictions where there is a legal requirement for testing new chemicals.
Another value that is frequently used is the NOEL or NOAEL, the No Observable (Adverse) Effect Level. The NOEL includes effects, such as minor weight loss, that are not considered to be adverse. These values are applicable only to that species in which the test was conducted. Extrapolation to other species will require dosage adjustment.
Probit Analysis
It is often desirable to compare the toxicity of one xenobiotic to that of another. This information may help to determine whether a substance used commercially or industrially can be replaced with a safer one, or whether a metabolite of a parent compound is more or less toxic than the compound itself. For this purpose, probit analysis is often used. When a toxic reaction is expressed as the number of experimental animals in a group displaying that reaction (e.g., kidney failure), the percent of a group responding to a given dose or exposure can be expressed as units of deviation from the mean. These are called normal equivalent deviations (NEDs). The NED for the group in which there were 50% responders would be zero, since it lies right on the mean. A NED of +1 corresponds to 84.1% responders. NEDs are positive or negative relative to the mean, so a value of 5 is added to each to make them all positive. The result is called a probit (for probability unit). Table 1.1 shows the equivalent probits and NEDs for given percent responses.
Conversion of Percent Responders to Probit Units
|
Percent Responders |
NEDa |
Probit |
|
0.1 |
−3 |
2 |
|
2.3 |
−2 |
3 |
|
15.9 |
−1 |
4 |
|
50.0 |
0 |
5 |
|
84.1 |
+1 |
6 |
|
97.7 |
+2 |
7 |
|
99.9 |
+3 |
8 |
a NED, normal equivalent deviation.
When quantal data are plotted as probit units against the log of the dose, a straight line results, regardless of whether the original data were distributed normally or were skewed. The method, in fact, assumes that the data were distributed normally. It is now easier to compare the quantal data for two different xenobiotics exhibiting the same toxic manifestation (or their lethality). These concepts apply equally to toxicological studies in mammals and in nonmammalian species. The following example illustrates these concepts using hypothetical toxicity data (Table 1.2) for two toxicants tested in fathead minnows (0.25–0.5 g). Each test group consisted of 100 fish. Values listed are mg/L concentration in water. Tables are available for conversion to probits.
Lethality in Fathead Minnows for Two Toxic Chemicals
|
Lethality (%) |
Fluorine (mg/L) |
Naphthalene (mg/L) |
|
10 |
25.0 |
0.5 |
|
20 |
50.0 |
1.0 |
|
60 |
100.0 |
2.0 |
|
84.1 |
200.0 |
4.0 |
When using aquatic or marine organisms for toxicity studies, it is important to remember that they are exposed to a given concentration of the test substance continuously, but they may not take it up instantly or even rapidly. A consistent time of exposure must therefore be incorporated into the experimental design. Figure 1.9, Figure 1.10, and Figure 1.11 illustrate arithmetic, semilogarithmic, and probit plots for these data.

Arithmetic plot of the lethality data shown in Table 1.2 for (a) naphthalene and (b) fluorine.

Semilogarithmic plot of % mortality vs. log of concentration. Semilog plot of data shown in Table 1.2 and in Figure 1.9 .
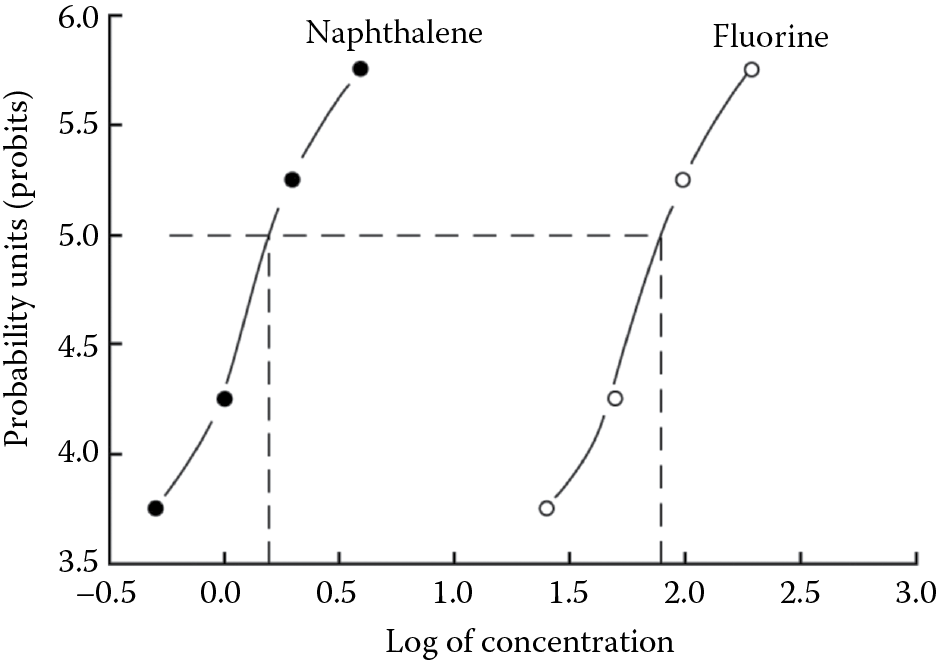
Probit plot of mortality for naphthalene and fuorine. Probits vs. log of concentration. Probit plot of the data shown in Table 1.2 and Figure 1.9 and Figure 1.10 .
Cumulative Effects
It may be that the manifestation of toxicity does not occur until the individual has been exposed continuously or repetitively (as with repeated, daily injections) for a prolonged period, perhaps days or weeks. This generally occurs with agents that are metabolized or eliminated very slowly, so that the rate of intake exceeds slightly the rate of detoxification and the drug slowly accumulates until a toxic level is reached. It is analogous to filling a bathtub with a faulty drain. The tub will fill, but only slowly because the water is running out almost as fast as it comes in. This involves the concept of biological half-life or T1/2. The plasma T1/2 of a drug or chemical is the time required for the plasma concentration to fall 50%. It is important to note that in most cases, this value is a constant for a given xenobiotic, i.e., it remains the same regardless of the initial level of the chemical. This is because the rates (pl) of biotransformation and excretion are usually concentration driven (the Law of Mass Action again), increasing or decreasing according to the plasma level. Biotransformations are enzymatic processes that generally obey first-order kinetics; i.e., the conversion rate is dependent upon the initial concentration of substrate. T1/2 values may also be established for other tissue compartments in the body. If the exposure interval greatly exceeds the T1/2 of a substance, it may be virtually eliminated between exposures. It requires about 5 × the plasma T1/2 to achieve virtual elimination. If the exposure interval is equal to or less than the T1/2, and the dose is constant, plasma steady-state concentration will be achieved in five plasma T1/2. The xenobiotic may be sequestered in organs and tissues. If no detoxification or elimination occurs, of course, the chemical will accumulate significantly with each exposure. Cumulative effects can occur as the result of repeated exposures even though no detectable levels of the toxicant accumulate, as when genetic damage is induced by carcinogens. Figure 1.12 illustrates the relationship of frequency of exposure and T1/2 with respect to clearance from tissues. These factors influence whether a toxic reaction is defined as acute (within 48 h), subacute (in 7–9 days), subchronic (±90 days), or chronic (>90 days).

Factors Influencing Responses to Xenobiotics
It should be evident that anything that influences the absorption, distribution, metabolism, or excretion of a xenobiotic will affect its toxicity. Many such factors exist.
Age
In general, biotransformation and excretion are less efficient at the extremes of life. Although drug metabolizing enzymes are detectable at mid-gestation, they do not become fully developed until 6–12 months of age. Thus, the toxicity of many substances is higher in neonates. An example is the antibiotic chloramphenicol, which can accumulate to toxic levels because the glucuronide-conjugase enzyme is lacking. Renal function also is underdeveloped so that excretion of drugs is impaired. The T1/2 for insulin clearance is 100 min in infants younger than 6 months of age versus 67 min in adults.
Body composition also differs with age. Total body water is 70%–75% of body weight in neonates versus 50%–55% in adults. Extracellular fluid is 40% of body weight in newborns versus 20% in adults. There is, thus, a greater fluid volume for dilution of water-soluble drugs in these infants. Their basal metabolic rate, moreover, is higher than in adults.
Other differences include greater permeability of skin, which has resulted in toxicity due to absorption of the hospital germicide hexachlorophene. Gastric pH is higher and gastric emptying is prolonged so that heavy metal absorption is increased. The intestinal flora will differ, and biotransformation by microbes will be different as well. In later childhood, these situations may be reversed because systems are functioning at peak efficiency.
After age 75, these same systems may have slowed down significantly compared to a 30 year old. Renal function and respiratory tidal volume are down, drug metabolizing enzymes are less efficient, and even the number of drug receptors may be lower. Body composition also changes. The ratio of fat to lean body mass increases with age and total body water is down. Cardiac output is reduced and perfusion is lower. Plasma albumin content is down. The toxicity of water-soluble toxicants like alcohol will be increased because it will be more concentrated. The concentration of the free component of albumin-bound agents will be increased because fewer albumin-binding sites are available. Biotransformation and excretion of xenobiotics will be impaired. The following are some examples of function at age 75 expressed as a percent of function at age 30 (Table 1.3).
Function at Age 75 as a Percent of Function at Age 30
|
Nerve conduction velocity |
90% |
|
Basal metabolic rate |
84% |
|
Cardiac output |
70% |
|
Glomerular filtration |
69% |
|
Respiratory function |
43% |
Body Composition
Body composition also varies considerably among normal individuals independent of age and that the factors discussed earlier with respect to fat and water content will be in play here as well.
Sex
Men and women may differ in response to xenobiotics. In part this is due to differences in body size, fat content, and basal metabolic rate. In one study, the T1/2 of antipyrine (an old and toxic analgesic) was 30% longer in young men than in young women. Differences in response to sex steroids obviously occur. Pregnancy is a special situation involving great changes in the metabolism, body composition, and fluid content of the mother. Placental transfer of many agents occurs and this may put the fetus at risk (see the following). Sex-related differences also occur in experimental animals. Cessation of respiration has been shown to occur more frequently in female than in male rats after barbiturate anesthesia.
Genetic Factors
For the majority of the population, biotransformations are controlled by multigenetic determinants. This is the basis of the continuous variation in response as reflected in the characteristic, normal population distribution curve. In some cases, however, a single gene locus may be responsible for altering the metabolism of a substance. This occurs in a subset of the population and affects a particular enzyme. The result is a discontinuous, bimodal distribution that reflects two, overlapping normal distribution curves.
In Figure 1.13, representative response distributions are shown for acetylsalicylic acid (ASA, aspirin) as an example of the multigene type of control and isoniazid as the single gene type, in this case for an acetylating enzyme.

Hypothetical dose–response distribution curves for a drug influenced by many genes (A) and one influenced by a single gene affecting drug biotransformation (B).
Pharmacogenetics is the subdiscipline of pharmacology that deals with this phenomenon. Over 100 examples have been identified of genetic differences in the biotransformation of drugs and chemicals. One of special concern for toxicology is the enzyme N-acetyltransferase. It acetylates and detoxifies many drugs and chemicals including the aryl amines that are potential carcinogens, as well as many drugs including isoniazid, an antitubercular agent, and sulfa drugs. Slow acetylation is the dominant pattern in most Scandinavians, Jews, and North African Caucasians. Fast acetylation predominates in Inuit and Japanese. Similar patterns have been found in other species including rabbits. Slow and fast oxidative metabolism has also been reported. About 9% of North Americans are slow metabolizers. In one study, a group of dye workers who were exposed to N-substituted aryl compounds were surveyed for the occurrence of in situ carcinoma of the bladder.
Those with the disease were predominantly slow acetylators, suggesting that slow acetylators probably accumulated a carcinogenic agent normally detoxified by N-acetylation.
Thiopurine S-methyltransferase (TPMT) also is subject to genetic polymorphism. This enzyme is responsible for S-methylation of the antineoplastic drugs azathioprine and 6-mercaptopurine. About 88.6% of humans have high, 11.1% intermediate, and 0.3% low or undetectable levels of enzyme activity. Individuals with high levels may be poor responders to cancer chemotherapy unless dosage is adequate, while those with low or undetectable levels are in danger of developing complete suppression of bone marrow function unless, again, dosage is adjusted downward. Other aromatic and heterocyclic sulfhydryl compounds are methylated by this class of enzyme, and there is little doubt that toxicity can be affected by genetic differences in enzyme activity. Similar genetic polymorphism has been demonstrated for the drugs debrisoquine and metoprolol, which are detoxified by hydroxylation. About 10% of male blacks develop hemolytic anemia when exposed to the antimalarial drug primaquine.
Pharmacogenomics is the science of using genotyping to identify individuals in advance who may pose therapeutic problems because of a lack or excess of detoxifying enzyme activity. It is now technically possible to do this through the use of modern techniques in molecular biology, notably DNA genotyping by the use of appropriate primers and the Polymerase Chain Reaction (PCR) method. Such technology may also be applied to the workplace to identify persons who are at excessive risk of a toxic reaction from a chemical in the work environment, or perhaps individuals who have already incurred DNA damage because of past exposure to a mutagen. These people could be excluded from high-risk areas (for them) or denied employment. Paradoxically, this possibility has already raised concerns in some union quarters that individuals might be denied their right to earn a living on biochemical grounds. In the future, employers might have to balance union concerns against the possibility of future litigation by workers made ill by their jobs. There is certainly no doubt that new ethical dilemmas will arise out of this technology.
Genetic factors also determine the emergence of strains of organisms resistant to normally toxic agents. There are many examples, including resistance of mosquitoes to DDT, rats to warfarin, malarial parasites to many drugs, cancer cells to anticancer drugs, and bacteria to antibiotics. In all cases, susceptible cells are killed off, leaving resistant mutants to proliferate. Mutations are occurring all the time. In bacterial populations, it has been estimated that a mutation imparting a degree of resistance to a drug occurs once in every 109 cell divisions. Unless that particular drug is present, however, the mutant strain will have no selection advantage and it will be overwhelmed by nonmutant cells. If the drug is present, however, it will have a distinct survival advantage and it will become the dominant form.
Genetic factors also may influence the response to a toxicant at the target site as well as at the site of biotransformation. Inherited disorders that render individuals more susceptible to drug-induced hemolytic anemia include glucose-6 phosphate dehydrogenase (G-6-PD) deficiency and sickle cell anemia. A host of drugs, including antimalarials and sulfonamides will induce a hemolytic attack in these people. There are also several inherited disorders of hemoglobin synthesis that act similarly. Many of the drugs involved are oxidizing, aromatic nitro compounds and nitrates. As noted earlier, altered gene coding plays a major role in carcinogenesis.
Presence of Pathology
Given that the liver and the kidneys are the major organs of detoxication, it follows that any serious impairment of their function will have a significant impact on the toxicity of xenobiotics. This has been observed in fatty necrosis of the liver, hepatitis, and cirrhosis. These will impact mostly on highly lipid-soluble agents requiring biotransformation to more water-soluble forms. Pharmacologically, this is seen with CNS depressants including the tranquillizers Librium and Valium. Kidney dysfunction is reflected mainly on water-soluble agents and their elimination may be greatly impaired by renal disease. Water-soluble antibiotics such as gentamicin have a greatly prolonged T1/2 in the presence of renal disease. Cardiovascular disease may affect tissue perfusion and the delivery of the xenobiotic to, or conversely, its removal from, its target site. Pulmonary disease will also affect the transfer of volatile agents across the alveolar membrane. In meningitis, the presence of inflammation compromises the integrity of the blood–brain barrier, and substances that would normally be excluded may reach significant concentrations in the spinal fluid. This fact makes some antibiotics (e.g., penicillin) useful to treat meningococcal meningitis even though they normally do not penetrate the barrier.
One of the most serious consequences of preexisting pathology may simply be the fact that if an organ has already lost much of its function, further damage by a toxicant may destroy it completely and create a life-threatening situation. This is one of the main reasons why the elderly are more vulnerable to toxic effects of drugs and chemicals.
Xenobiotic Interactions
The effect that one drug may have on the action of another, collectively known as drug interactions (or drug–drug interactions), is an important aspect of clinical pharmacology. The effects of two drugs given together may be additive if they induce the same response (even through different mechanisms), synergistic if the total response of their combined effect is greater than the predicted sum of their individual effects, or antagonistic if one drug diminishes or prevents the effect of the other.
Mechanisms involved in drug interactions include altered absorption from the gastrointestinal tract, altered excretion (renal, biliary, respiratory), competition for receptors (antagonism), summation of pharmacological effects, and altered biotransformation. Drug interactions are usually associated with drug adverse reactions, but they may also be exploited. All antidotal remedies are based on drug or chemical interactions. Examples of antidotes include the use of atropine to treat organophosphorus poisoning, metal chelators for treating lead and other heavy metal poisoning, the use of activated charcoal to bind toxicants in the gastrointestinal tract and prevent their absorption, emetics to induce vomiting and the removal of toxicants, naloxone to reverse the respiratory depression of opiates, and N-acetylcysteine to treat acetaminophen poisoning.
When more than two substances are present, the possibility for a drug interaction is much greater. This is of special concern in the area of environmental toxicology because of the multiplicity of xenobiotics that may be present in water, food, and air. Most of the attention has centered on the possibility that the presence of one substance may increase the concentration of carcinogens from other sources by affecting their metabolism through enzyme induction. We have already discussed the effect of cigarette smoke on hepatic enzymes. Many volatile solvents also are enzyme inducers, and chronic exposure to low levels of these, as in the industrial setting, can lead to increased enzyme activity. It has been shown experimentally that bedding rodents on soft-wood shavings induces hepatic microsomal enzyme activity because of the volatile terpenes given off. Workers in soft-wood sawmills could experience similar effects. Literally hundreds of industrial chemicals have been shown to be enzyme inducers, including benzo[a]pyrene and 3-methylcholanthrene. Others include insecticides, (DDT, aldrin, dieldrin, lindane, chlordane), PCBs, polybrominated biphenyls (PBBs), dioxin, and drugs such as phenobarbital and other barbiturates, steroids, and others. Even though a substance is itself not toxic at a particular exposure level, it may influence the toxicity of other agents. The enzymes usually involved are the CYP450 monooxygenases, but conjugating enzymes also may be induced. Food may contain natural enzyme inducers. Potato contains “a-solanin,” and tomato “tomatin,” both of which are steroidal alkaloids. The bioflavonoids are fairly potent inducers. They are found in species of Brassica including Brussels sprouts, where they have been shown to affect the metabolism of some drugs. Rutin is a bioflavonoid found in buckwheat in fairly large amounts. While it is difficult to identify situations where these various inducers have influenced toxicity (with the exception of cigarette smoke), their ubiquity illustrates how different individuals or groups may display different sensitivities to the same toxic exposure on different occasions. More recently, it has been shown that fruit juices, especially grapefruit juice, can have a significant impact on the metabolism of some drugs. Naringin is a flavonoid in grapefruit juice and hesperidin, a flavonoid in orange juice. Both have been shown to inhibit enteric CYP3A4, which is a barrier protein for the absorption of many drugs. They also inhibit the organic anion-transporting polypeptide (OTAP) 1A2 and are known to interfere with the absorption of many drugs. The list of food–drug interactions grows longer year by year.
Organic solvents have been shown to influence ethanol toxicity. Toluene depresses alcohol dehydrogenase and prolongs the ethanol T1/2. Ethanol itself may increase the liver and CNS toxicity of CCl4, trichloroethylene, and others.
Diet can also affect the toxicity of substances in other ways. A toxicant may be adsorbed to dietary components that reduce its absorption. The ability of dietary calcium to reduce lead toxicity is well documented. Lead and calcium appear to compete for the same absorption site on the intestinal mucosa so that a diet high in calcium will reduce lead absorption. A diet high in fiber will shorten the transit time of the gastrointestinal tract so there is less time available for absorption. This may be one reason why high fiber diet is associated with a lower incidence of colon cancer.
Some Toxicological Considerations
Acute versus Chronic Toxicity
Acute and chronic toxicity for a single agent may be quite different and one is not a reliable predictor of the other. For example, acute benzene intoxication involves CNS disturbances such as excitation, confusion, stupor, and convulsions. Chronic toxicity includes depression of the bone marrow and a reduction of all circulating blood cells (pancytopenia) and benzene is carcinogenic in experimental animals. Chronic carbon monoxide (CO) poisoning is experienced by heavy smokers who may suffer from headache, dizziness, and shortness of breath. Acute CO poisoning affects firemen and fire victims who may become comatose. Numerous other examples exist. A major area illustrating this is that of tumor formation. A short-term exposure to a substance may elicit acute toxicity without significant risk of carcinogenesis, whereas long-term exposure to very low levels may not result in any toxic manifestation but could induce tumor formation. Dioxin (TCDD) is a prime example of this. Acute exposure causes the skin rash known as chloracne, whereas long-term exposure may be carcinogenic.
A detailed discussion of toxic mechanisms is beyond the scope of this chapter. More detailed descriptions are given with the discussions of specific chemicals and groups of chemicals. What follows is a brief overview to illustrate the role of target organs and systems in toxic reactions.
Acute Toxicity
Acute toxicity generally refers to effects that occur following a 24–72 h exposure to a single or multiple doses of a toxicant. Effects are usually observed within a few days. If the agent is rapidly absorbed, the effect may be immediate. The CNS is very vulnerable to acute toxicity from very-lipid-soluble agents.
Peripheral Neurotoxins
Organophosphorus and carbamate pesticides inhibit acetylcholinesterase (AChE) so that acetylcholine (ACh) accumulates and overstimulation of receptors occurs. ACh is a neurotransmitter in both the central and peripheral nervous systems. There are many naturally occurring neurotoxins, including tubocurarine, which blocks nerve transmission to voluntary muscle; botulinum toxin (from Clostridium botulinum), which prevents the release of ACh from nerve endings; and tetrodotoxin (from the puffer fish), which paralyses nerves by blocking sodium channels. Belladonna alkaloids (atropine and scopolamine) from nightshade are muscarinic blockers with peripheral and central effects and muscarine from mushrooms is a muscarinic stimulant. Nerve gases are also irreversible AchE inhibitors. Many other examples exist (see Chapter 11).
Central Neurotoxins
The inhalation of many volatile organic solvents and petroleum distillates can act like anesthetics and may cause unconsciousness. This has occurred in industrial accidents and in substance abuse (e.g., gasoline sniffing). Ethyl, methyl, and isopropyl alcohols are CNS depressants.
Inhibitors of Oxidative Phosphorylation
Cyanide (CN) in the form of cyanogenic glycosides (e.g., amygdalin) is present in many plant components including almonds, the pits of cherries, apples and peaches, plums, apricots, and wild chokecherries. Human poisonings have occurred from consuming too many of these seeds, and livestock are often poisoned from eating chokecherry bushes. Cyanide binds to heme to prevent electron transfer. Cyanide may be present in metal ores, in some pesticides, and in metal polishes. The tragic accident at Bhopal, India, which killed over 2000 people, was due to the release of 40 tons of methyl isocyanate from an American Cyanamid plant. Azide and hydrogen sulfide act like CN.
Carbon monoxide (CO) combines with hemoglobin and cellular cytochromes and prevents association with O2. It thus causes cell hypoxia and also interferes with O2 transport by red blood cells.
Uncoupling Agents
Many agents act as uncouplers, preventing the phosphorylation of ADP to ATP, the high energy phosphate. Uncouplers include the herbicide 2,4-D, halogenated phenols, nitrophenols, and arsenate. Most contain an aromatic ring structure. O2 consumption and heat production are increased without an increase in available energy.
Inhibitors of Intermediary Metabolism
Certain fluoroacetate compounds of natural origin are used professionally as rat poisons. They inhibit the citric acid cycle to deplete available energy stores. The heart and the CNS are the organs of toxicity.
Chronic Toxicity
Exposure to some toxicants must occur over days, weeks, or months before signs of toxicity appear. Heavy metals tend to act in this manner. Several outbreaks of methylmercury poisoning have followed this pattern. Mercury (Hg) from industrial discharges may be converted to methylmercury by microorganisms in the water. Monomethylmercury is CH3Hg and dimethylmercury is (CH3)2Hg. These accumulate up the food chain to concentrate in fish and shellfish that may be consumed as food. As the toxicant accumulates in the tissues, severe neurological disorders occur. This happened at Minamata Bay in Japan, and on the Grassy Narrows reserve in Northern Ontario. Infants born to exposed mothers may suffer from a cerebral palsy-like syndrome.
Cadmium (Cd), used in nickel–cadmium batteries, in electroplating, and in pigments, may accumulate in workers and cause kidney damage. Similar cases in the general population have been reported in Japan, from eating rice and other grains grown in soil contaminated with industrial wastes. Carbon tetrachloride (CCl4) was used extensively in the dry-cleaning industry before it was discovered that is caused hepatic necrosis because it was activated to a free radical by CYP-450-dependent monooxygenase.
Mutagenesis and Carcinogenesis
Introduction
Sir Percival Pott was a senior surgeon at St. Bartholomew’s Hospital in London, England, in the latter half of the eighteenth century. He is widely credited with being the first person to make a connection between exposure to a foreign substance and the development of a cancer. In 1775, he noted a high incidence of scrotal cancer, subsequently shown to be squamous cell carcinoma, in chimney sweeps. The common term for this condition, soot wart, suggests that workers in the trade had already made the connection. Boys as young as four were employed as chimney sweeps, often pressed into service by unscrupulous adults. In 1788, the Chimney Sweepers Act was passed by the British Parliament, becoming the first legislation designed to eliminate child labor, at least in this trade.
Cancer will afflict one in four North Americans at some point in their life and will be responsible for the death of about one in five of them. While chemicals are responsible for many cancers (this is the cause that creates the most concern among the populace), viruses and radiation, either natural or from anthropogenic sources, are also important causes. Cancer can be described as an aggressive and inappropriate growth of a cell type in the body. If the cell type remains true to its origin and can always be identified as that cell type, the tumor is generally benign and does not metastasize beyond the primary site. It can still become excessively large and interfere with normal bodily function if not removed. If the cancer cells lose their defining characteristics and become undifferentiated, the tumor is malignant and will most likely metastasize to distant sites if it is not treated successfully by surgery and/or antineoplastic drugs.
A characteristic of chemically induced carcinogenesis is that it is believed usually to involve decades-long exposure to very low levels of carcinogens, making predictions from animal studies very difficult. This is clearly illustrated by the association of cigarette smoking and lung cancer, which is discussed in Chapter 4. An exception is the process of chromothripsis (see Model 6 given in the following). A wide range of chemically diverse, natural and synthetic chemicals may induce alterations in DNA that, depending on the nature of the defect and the timing of its occurrence, can cause a neoplasm, a heritable change (mutation), or a developmental birth defect. It should be noted that birth defects (teratogenesis) may result from chemical interference with many other cell processes not involving altered DNA, such as interference with essential substrates and precursors, impaired mitosis, enzyme inhibition, altered membrane characteristics, etc. Most anticancer drugs are teratogenic for the same reason they are antineoplastic. Mutations are not always harmful, and they provide the necessary genetic diversity for natural selection to occur, but they can also be responsible for fertility disorders, hereditary diseases, cancers, and malformations. Three types of genetic abnormalities may be induced.
Chemically induced carcinogenesis differs from other manifestations of toxicity in that the usual exposure–response relationship does not generally apply. The xenobiotic or its metabolite attaches irreversibly by binding to an essential component of the living cell to disrupt normal function. The binding sites are usually on macromolecules such as nucleotides, nucleosides, regulatory proteins, RNA, and DNA. The effects may be cumulative, and they cannot be related to blood or tissue levels at the time of their manifestation. There may be a considerable delay from the initial exposure to the emergence of toxicity. This kind of reaction is involved in mutagenic and carcinogenic effects, and also causes a type of anemia, called aplastic anemia, in which the capacity of the bone marrow to produce blood cells is permanently destroyed. Some older drugs such as chloramphenicol and phenylbutazone have caused aplastic anemia in a very small number of patients; perhaps one in 40,000 exposed persons. The solvent benzene, a carcinogen, has also been associated with aplastic anemia. These low frequency toxicities are difficult to identify and necessitate careful risk/benefit analysis when therapeutic interventions are contemplated.
Our understanding of the carcinogenic process has been greatly enhanced by the development of DNA technology and the mapping of the human genome. Nonetheless, it is a highly complex, even confusing, process that is in a continual state of flux. What follows is an attempt to organize and simplify the story. It should not be viewed as a fully detailed, complete description of the process.
Genetics of Carcinogenesis
Oncogene
This is a gene that predisposes a normal cell to become a malignant one. There are scores of known human oncogenes with more being discovered all the time. Oncogenes can be turned on by external factors such as radiation, viruses, chemicals that damage DNA throughout life, or by inherited factors actors. Unlike normal genes, they cannot be turned off but remain in a state of constant activity. The first oncogene to be identified was a component of a cancer-causing virus in chickens, the Rous sarcoma, named after the discoverer. Labeled the src (sarc) oncogene, it has been identified in humans and is associated with colon, liver, lung, breast, and pancreatic cancer. It has since been shown that viral oncogenes like src are not part of the normal genetic complement of the virus but are acquired from normal cells by the retrovirus. Retroviruses may cause the excessive production of an oncogene, induce its expression in an inappropriate cell, or mutate the coding of a gene during transduction. Human tumor oncogenes have been identified as alleles of the ras family of proto-oncogenes. Others include fos, jun, and myc. The myc oncogene was first identified as a viral one and is important in human tumorigenesis as a growth factor stimulant. Cellular oncogenes are designated by the prefix “c” (e.g., c-myc, c-abl). Viral oncogenes are designated by the prefix “v.” The genetic mutations BRCA1 and BRCA2 are predisposing risk factors for breast cancer.
Proto-Oncogene
A proto-oncogene is the normal counterpart of an oncogene. Proto-oncogenes are normal constituents of the cell that exist in every cell component (membrane, cytoplasm, nucleus, etc.) and that are involved in every signal transduction cascade. They modulate cell function, growth, and proliferation. When acted upon by external influences they are converted to oncogenes, resulting in uncontrolled cell growth. If a proto-oncogene undergoes a somatic mutation, regulation of growth in that cell is lost and cancer can occur. This can be triggered by a number of molecular mechanisms.
Tumor Suppressor Genes
These recessive genes are part of the normal human genome and are capable of preventing the onset of cancer even in the face of the molecular events noted earlier. They are sometimes referred to as “anti-oncogenes.” They were discovered as a result of the observation that when normal cells are fused with tumor cells in culture, suppression of the tumor cells resulted. Inactivation of these suppresser genes may be an essential step in tumorigenesis, at least in some cases, and prevention of this inactivation by inserting normal copies of tumor suppresser genes, or by mimicking their function pharmacologically, may lead to new therapies. Tumor suppressor genes (TSGs) may be responsible for the existence of “cancer families.” If an inherited defect in one copy acquires a matching defect in the other copy through retroviral or chemical damage, or from the other parent, cancer may result. Much has been learned about TSGs from the study of a juvenile eye cancer, retinoblastoma. This is caused by a mutation in the Rb gene located on chromosome 13. It normally suppresses the tumor in its dominant phenotype. The mutation must be present in both alleles for the tumor to develop. The retinoblastoma story is rather complex and it serves to illustrate the complicated nature of carcinogenesis. The product of the normal Rb gene interacts with a protein, E2F, a nuclear transcription factor involved in cellular replication functions during the S phase of the cell cycle. The interaction with the Rb product prevents this function, inhibiting mitosis. The mutant form of Rb cannot interact with E2F, so cell growth is not inhibited. The retinoblastoma story is discussed further under the “Two-hit” model.
Another TSG is p53 located on chromosome 17. It is a protein containing 393 amino acids, and a substitution of a single one can lead to loss of function. Both recessive and dominant mutations can occur. Other TSGs include Rb, involved in retinoblastoma and osteosarcoma and APC, involved in colon cancer. E-cadherin, a Ca++-dependent transmembrane protein involved in epithelial cell–cell interactions, has been suggested as a candidate TSG.
Growth Factor Receptors
Many tumors may express genes for receptors that are receptive to agents that promote the aggressive growth of the tumor. The epidermal growth factor receptor is one such. It is known by many names such as EGFR, ErbB-1, and, in humans, HER-1. It is one of a family of receptors known as the ErB family. Others are HER2/c-neu (ErbB2), HER 3 (ErbB-3), and HER 4 (ErbB-4). These receptors bind to specific ligands like epidermal growth factor and transforming growth factor α (TGFα). The result may be accelerated tumor growth through uncontrolled cell division.
Many tumors have receptor HER2 including breast cancer, colon cancer, lung, and likely squamous cell carcinoma of the head and neck, and probably many others. New drugs that alter the receptor affinity for HER2 can be useful in suppressing tumor growth. These new drugs include trastuzumab (Herceptin®) and cetuximab (Erbitux®).
Hormone Receptors
The presence of cytoplasmic receptors for estrogen and progesterone is a critical factor in breast cancer and determines the nature of the therapy. Although it may seem paradoxical, patients whose primary tumor is receptor-positive generally have a more favorable course than those whose tumors are receptor-negative. This is because it is possible to manipulate the hormonal environment with receptor-blocking drugs. Tamoxifen is an estrogen receptor modulating agent that is a first-line drug in treating patients with estrogen receptor-positive breast cancer.
A difficult form of breast cancer to treat is the so-called triple “negative” breast cancer. This cancer lacks receptors for estrogen, progestin, and the epidermal growth factor (receptor HER2). They are thus not responsive to treatment with receptor blocking agents like tamoxifen for estrogen receptors or Herceptin for HER2 receptors but the oncologist must rely on other treatments like radiation and conventional chemotherapy.
Hormonal manipulation is also employed in the treatment of metastasized carcinoma of the prostate gland in men. These tumors have cytoplasmic androgen receptors that are responsive to androgens like testosterone (which is actually converted to dihydrotestosterone) that promote proliferation of the tumor. Antiandrogen therapy is used to retard this. Older techniques included removal of the testes and the use of the synthetic estrogen diethylstilbestrol. Flutamide and bicalutamide are androgen receptor antagonists. A family of drugs that includes leuprolide (Lupron) and goserelin (Zoladex) are analogs of gonadotropin-releasing hormone (GnRH). There is a complex feedback loop controlling the synthesis of androgens and estrogens. The hormone GnRH (also known as LHRH for luteinizing hormone-releasing hormone) is synthesized and released from the hypothalamus in a pulsatile fashion. Low frequency pulses stimulate the release of follicle-stimulating hormone (FSH) and high frequency pulses the release of luteinizing hormone (LH), both from the anterior pituitary gland. Synthesis and release of FSH and LH are also affected by the levels of circulating estrogens and androgens in a negative feedback manner.
Analogs of GnRH occupy GnRH receptors on the anterior pituitary, initially causing increases of FSH and LH until the receptors become desensitized and levels of FSH and LH begin to fall, as do levels of androgens. Circulating levels can reach those associated with castration and the name chemical castration has been applied to this hormonally based approach.
Endocrine-disrupting chemicals, both natural and synthetic, may also play a role in the development of mammary tumors. These will be discussed in Chapter 12.
Drug Resistance Genes
It is well known that cancer cells can develop resistance to antineoplastic drugs just as bacteria become resistant to antibiotics and rats to warfarin. The T790M mutation and MET oncogene impart resistance to drugs targeting the EGF receptors. Natural EGFR inhibitors also exist, one being a potato carboxypeptidase inhibitor. A multidrug-resistant gene, MDR-1, encodes for a P-glycoprotein that pumps cytotoxic, antineoplastic drugs out of tumor cells.
Antisense Genes
An even more recent development in the genetic manipulation of tumor cells is the synthesis of “antisense” sequences. These oligonucleotides are short (15–25 bases), single-stranded, DNA sequences that have been altered so that they target a specific mRNA sequence, resulting in a defective DNA segment in a highly specific manner. Thus, if a specific segment (sense) that plays a key role in tumorigenesis can be identified, the appropriate antisense sequence can be formulated and inserted to interfere in the process. This advance holds great promise for the future treatment of cancer.
The World Health Organization’s International Agency for Research on Cancer maintains an excellent website with evaluations on the current carcinogenicity rating of hundreds of chemicals (http://www.iarc.fr/).
Genetic Predisposition to Cancer
Just how much cancer is due to environmental factors and how much is due to a genetic predisposition has been a matter of considerable debate. Recently, a study conducted by the Department of Medical Epidemiology at the Karolinska Institute has shed some light on this question. They combined data from 44,788 pairs of twins listed in the Swedish, Danish, and Finnish twin registries to examine the risk of cancer for persons with a twin with cancer at one of 28 anatomical sites. Statistically significant evidence of effects of heritable factors was observed for several cancers. Analysis indicated that genetic predisposition accounted for 27% of the risk for breast cancer, 35% of the risk for colorectal cancer, and 42% of the risk for prostate cancer. Overall, the risk of being diagnosed with any of these cancers by age 75 ranged from 11% to 18% for a person whose identical twin had that cancer and from 3% to 9% for a person whose fraternal twin had that cancer. The authors concluded that environmental factors were by far the greatest contributors to the risk of getting cancer.
Epigenetic Mechanisms of Carcinogenesis
Although it must be stated that no malignancy progresses without some of the genetic factors discussed earlier, carcinogens that function through non-DNA-reactive processes have been identified in recent years. They do not induce mutation in cell assays nor do they induce direct DNA damage in target organs. Precise mechanisms by which these agents operate have not been established but changes in gene expression and cell growth (proliferation) seem to be central to the process. Some may be tumor promoters that cause proliferation of initiated cells. Phorbol esters of plant origin seem to act this way. Cytotoxic agents, by killing large number of cells, trigger the production and release of growth factors, and the frequency of mutations is thus also increased. Some of these may be malignant cell types. Chloroform, by inducing liver necrosis and hyperplasia, can induce tumors in this way. Phenobarbital, conversely, can induce mitosis without cytotoxicity by mechanisms that are not clear. Rapid cell division can lead to error-prone repair and hence mutation. This relates to the Ames hypothesis discussed later. Under the influence of mitogenic factors, a mutant cell may proliferate and become the dominant cell type.
Another mechanism that may operate for nongenotoxic carcinogens is interference with normal apoptosis. Apoptosis, or programmed cell death, is a normal process by which senescent cells or cells with massive DNA damage are removed. Cancer results from an imbalance between cell growth and cell death with resulting unchecked proliferation. Several tumor promoters have been shown to inhibit normal apoptosis. The process of apoptosis involves oncogenes, TSGs, and cell cycle regulatory genes, and interference with these can lead to cell proliferation and neoplasia. Obviously there are genetic influences at work here as well.
Many of these processes may be facilitated by interference with cell–cell communication across gap junctions. Gap junctions are areas of cell–cell close contact containing channels that allow the passage of ions and small water-soluble molecules. Signal-transducing substances such as calcium, inositol trisphosphate, and cyclic AMP are transmitted in this way. Gap junction communication has been shown to be decreased in the presence of some epigenetic carcinogens.
Induction of cytochrome P450 may result in the formation of reactive intermediates that can act as promoters, often through the formation of free radicals.
Viruses and Cancer
The story of the Rous chicken sarcoma virus and its role in oncogene formation has been dealt with earlier. The list of viruses that can play a role in carcinogenesis is growing quite long and no doubt will continue to do so. The list includes Epstein–Barr virus (EBV), human papilloma virus (HPV), hepatitis C virus (HCV), hepatitis B virus (HBV), human herpes virus 8 (HHV-8), Human T-cell leukemia virus type 1 (HTVL-1).
The Rous sarcoma virus is a retrovirus. Retroviruses are a major factor in the conversion of proto-oncogenes to oncogenes. These viruses contain RNA rather than DNA and redirect host cell to produce viral DNA through the process of reverse transcription. This involves an enzyme, “reverse transcriptase.” Proto-oncogenes may be mutated to oncogenes during this process. Over 30 human oncogenes have been identified to date. In general, tumor viruses have the ability to integrate into the host genome and express their viral oncogenic proteins. Prior exposure to EBV and HPV has become a recognized risk factor for many cancers.
Viruses are also being employed as therapeutic agents in cancer therapy. Viruses can be reprogrammed into oncolytic vectors. Several modifications are employed. Targeting introduces multiple layers of cancer specificity, improving safety and efficiency. Arming refers to the expression of pro-drug convertases and cytokines. Shielding refers to coating the virus with polymers and the use of different envelopes (capsids) to protect the virus from the host’s immune response. While gene therapy has many potential therapeutic uses, over half of currently approved gene therapies relate to the treatment of cancers. The following lists ways in which such therapy is employed.
Inactivation of oncogenes: Oncogenic proteins are associated with many malignancies and blocking their expression would be a desirable target in cancer therapy. The adenoviral E1A interferes with the transcription of the oncogenic protein erb-2, common in breast and ovarian cancer.
Augmenting TSGs: Mutations in the numerous TSGs are the factor in the development of a neoplasm. Using adenoviral vectors some success has been obtained by introducing the TSG p53. BRCA1 and BRCA2 (standing for breast cancer susceptibility genes 1 and 2) are TSGs. A mutation of one of these genes is a significant risk factor for breast and ovarian cancer and these mutations can be heritable. A viral vector has been used to introduce normal BRCA1 into ovarian cancer in an attempt to induce suppression of the tumor with some success. Some cases fail because the mutant gene can be dominant and prevent the normal gene from acting.
Induced cell death: The inclusion into a virus of a gene that promotes the conversion of an antineoplastic pro-drug to its active form can, when given in combination with that drug, cause lethal concentrations of the drug to accumulate in the tumor cell. This is accomplished by using a pro-drug-converting enzyme such as cytosine deaminase for 5-fluorocytidine, or deoxycytidine kinase for fludarabine and 2-chlorodeoxyadenosine.
Chemoprotection: There is a protein known as P-glycoprotein that is capable of transporting or “pumping” numerous antineoplastic drugs out of tumor cells, thus preventing the drugs from reaching lethal concentrations. This form of multidrug resistance (MDR) can be a thorny problem for cancer chemotherapy. The MDR-1 gene encodes for the P-glycoprotein. In a departure from the usual gene therapy approach, the MDR-1 gene in a retroviral vector is being studied to protect normal bone marrow cells from the cytotoxic effects of antineoplastic agents used to treat tumors elsewhere. Because of their high rate of cell division, bone marrow cells are very vulnerable to cytotoxic effects.
Viral-mediated oncolysis: Some viruses, notably adenovirus and HSV-1, are capable of lysing tumor cells that they infect. When the tumor cells lyse, the virus is released to infect adjacent cells. Using such viruses in combination with other therapies, including other gene therapies, is showing promise for cancer treatment.
Immunomodulation: Tumor cells generally have impaired immune function. Cytokines may enhance immunity against cancer cells. Genetically engineered tumor cells have been shown experimentally to be less able to establish tumors in vivo. Infecting tumors with these cytokine-producing genes may increase their vulnerability to the host immune system, especially if used in conjunction with immunostimulatory agents that do not affect tumor immunity.
The search for new ways to exploit knowledge of tumor genetics for therapeutic gain continues and will doubtlessly lead to novel and hopefully successful approaches.
Models of Carcinogenesis
An interesting article by Vineis et al. discusses the changes in our understanding of carcinogenesis and how it has evolved over time. They note that six alterations in cell physiology are required to initiate and perpetuate malignant growth. These are as follows: (1) self-sufficiently in growth signals; (2) insensitivity to inhibitory (antigrowth) signals; (3) avoidance of programmed cell death (apoptosis); (4) unlimited replicative potential; (5) sustained angiogenesis (development of new blood vessels); (6) tissue invasion and metastasis. They also propose several models of carcinogenesis. They chose the term model rather than theory because there is experimental and mathematical evidence in support of them. While conceding that their approach is largely artificial they find it useful to promote discussion and understanding.
Model 1
The “mutational” model places emphasis on point mutation and chemical carcinogens. Mutation is the main change that leads to a malignant phenotype. The mutation becomes fixed so that the progeny of the mutated cell is abnormal. (Spontaneous natural mutations also occur, and this is the basis of natural selection. If a mutation provides a survival advantage to the individual it may become a dominant feature of the species. If it is disadvantageous it likely will not survive.) Point mutation, also called gene-locus mutation, involves the alteration in some way of a small number of base pairs of nucleic acids. This may involve deletion, addition, or the substitution of an incorrect base pair. Strictly speaking, the term mutagenesis refers exclusively to this type of DNA alteration.
The discovery of the carcinogenicity of tobacco smoke and of PAHs contributed to the formulation of this model. Research into the identification of reaction products between chemicals like PAHs and macromolecules like DNA adducts contributed to the development of the model as did the study of induced mutations in bacteria. Viral research also played a role (see src oncogene discussed earlier).
Bruce Ames, in his 80s (as this is written), is Professor of Biochemistry and Molecular Biology at the University of California in Berkeley. He is well known for the development of the Ames test, a bacterial culture test widely used for the identification of mutagenic chemicals. It is widely recognized that the frequency of mutagenesis increases with the frequency of cell divisions (mitogenesis) and thus the likelihood of a neoplastic mutation also increases as well. Ames postulated that the standard animal tests for detecting chemical carcinogens, that employ very high doses to elicit a measurable response, were producing an inordinately high rate of false positives. Very high doses of many, if not all, chemicals are cytotoxic, and when large numbers of cell are killed it stimulates mitosis. The development of cancer, therefore, could be a nonspecific response to the high rate of mutagenesis. Ames also postulated that endogenous mutations were a major contributor to the aging process. Prof. Ames’ website may be viewed at http://www.bruceames.org. Ames incurred the wrath of environmentalists and some fellow scientists when he declared that our obsession with eliminating trace levels of chemicals, notably pesticides, from our diet was misplaced. He based his premise on the knowledge that we consume hundreds, if not thousands, of natural pesticides that have never been tested for carcinogenicity or mutagenicity and the few synthetic ones to which we are exposed are inconsequential by comparison. While his theory remains largely untested, it constitutes an important reminder that our natural environment is not without risks and that we are in possession of protective mechanisms that have evolved against them, as is discussed later. Some perspective is often required in judging relative risks whether from anthropogenic or natural sources.
Model 2
The “genome instability” model evolved from research on familial cancers. The study of retinoblastoma was the basis for the “two-hit” hypothesis of cancer development and the (then) theory of TSGs. The discovery of Rb1, the TSG that leads to retinoblastoma when both copies of the gene are mutated, confirmed the theory. In this case, one hit was the inherited predisposition for the tumor and the second hit was the deactivation of the TSG. A double hit could also involve growth promotion of the tumor cells combined with deactivation of a TSG. A related, recently described model called chromothripsis is discussed later.
Model 3
The “nongenotoxic” model is characterized by emphasis on nongenotoxic effects of cancer risk factors that do not act through DNA alterations. These would include such risk factors as diet, obesity, hormones, and insulin resistance.
Model 4
The “Darwinian” model applies Darwin’s theory of natural selection at the cellular level. Cells with a greater propensity to divide may have a selection advantage and may through other processes mutate to become malignant cells.
Model 5
Vineis and his group note that more recent models put emphasis on the microenvironment surrounding the cancer cells that may have been involved in creating favorable conditions for their development.
Model 6
A new model has been added since the paper by Vineis’ group. Chromothripsis is the name applied to a new form of carcinogenesis. In January of 2011, a paper appeared in the journal Cell describing how a catastrophic event could shatter a chromosome into myriad pieces, which might then be reassembled incorrectly, producing numerous mutants that could conspire to cause a multihit form of carcinogenesis. The conventional theory that cancer always required many years to develop was thus also shattered as this form of cancer could appear without needing any preexisting genetic predisposition or concomitant risk factors. What might cause such a catastrophic event remains largely a mystery, although radiation is thought to be one possibility. It has been observed that bone cancer sometimes develops suddenly in persons exposed to radiotherapy. Two to three percent of all cancers and 25% of bone cancers appear to result from such a chromosome-shattering event. It seems highly likely that the story of carcinogenesis is not yet finished.
Stages of Chemically Induced Carcinogenesis
The induction of a cancer is a complex, multistage process involving interactions among the carcinogen, environmental, and endogenous factors. Three stages are generally recognized: (1) initiation, (2) promotion, and (3) progression. It should be noted that the description of these stages is largely one of observed behavior rather than any genetic or biochemical characteristic.
Initiation
The induction of a mutation by an electrophilic chemical or metabolite (a genotoxic carcinogen) that binds to DNA is believed to be the initial step. DNA thus altered is called an adduct. The heritable characteristic is not expressed. Chemicals that are capable of inducing tumors after a single exposure are sometimes called complete carcinogens (see also Model 6). This is usually seen in animal studies. PAHs such as dimethylbenz[a]anthracene and 3-methylcholanthrene are initiators.
Promotion
Promoters are agents that increase the number of tumors, increase their growth rate, or decrease the latency period. They do not bind to DNA and are, therefore, called epigenetic carcinogens. Exposure to the initiator must occur first. Promoters do not themselves generally cause tumors, although there is evidence that some promoters are merely weak carcinogens that act synergistically with other carcinogens. Co-carcinogens are agents that, when present just before, or together with, a carcinogen, result in significantly higher tumor yields in experimental animals some substances may be both promoters and co-carcinogens (e.g., phorbol esters). Nutritional factors (e.g., saturated fat intake), hormones, trauma, and viruses may act as co-carcinogens.
Progression
This refers to the natural history of the disease. At this stage there are irreversible changes resulting from mutation in one or more genes controlling key aspects of cell regulation. This instability of the karyotype increases with tumor growth. Some chemical carcinogens act at this stage, including arsenic salts, benzene, and asbestos. Figure 1.14 summarizes the steps involved in carcinogenesis.

Given the large number of chemicals that are carcinogenic, and the number coming onstream daily, testing for carcinogenicity is of paramount importance and of some difficulty. This subject will be dealt with in Chapter 2.
DNA and Cell Repair
Response of Tissues to Chemical Insult
Tissue regeneration occurs after destruction of some cells. This may lead to the release of growth factors that stimulate cell division and proliferation, followed by maturation of the cells to a functional state. Function is thus restored. Cell regeneration may occur if a portion of a cell is destroyed but the nucleus remains intact. Thus, the axon of a neuron may eventually regenerate if the cell body is undamaged.
Hyperplasia refers to an increase in the size of an organ or tissue in response to increased demands upon it. This is a normal adaptive phenomenon and it involves an increased number of normal cells. It tends to occur in organs of intermediate specialization such as the liver, pancreas, thyroid, adrenal cortex, and ovary. Hyperplasia of the liver may occur in response to an increased demand to detoxify a xenobiotic presented in high concentration or for a long period.
DNA Repair
DNA becomes damaged as a result of everyday exposure to toxic substances, the generation of free radicals, exposure to natural source radiation, etc. It is therefore not surprising that enzymatic DNA repair mechanisms exist. The characteristic of self-repair appears to be unique to the DNA molecule. Both constitutive (ongoing) and inducible (activated in response to an event) systems exist. The natural, endogenous repair system can be overwhelmed by massive or repeated exposure to genotoxic agents. In that case, primary DNA damage occurs (strand breaks, loss of bases, or adduct formation).
The ability to remove and replace damaged segments of DNA is central to the repair process. If repair can be accomplished prior to the fixation of the mutation, there may be no adverse affect from the DNA damage. DNA damage may inactivate both error-prone and error-free processes. Error-prone processes may actually lead to new mutations through nucleotide mismatches. Error-free processes will correct the damaged DNA site with the correct nucleotide sequence. Which process is dominant depends on a number of factors including species, (processes tend to be species-specific), cell type, the nature of the chemical mutagen, and the nature of the lesion. 3′ and 5′ endonucleases cleave the DNA on either side of the adduct and an exonuclease cuts out the damaged region, including nucleotides on either side of it. A DNA polymerase inserts the correct bases into the patch and a DNA ligase seals it.
Even in error-free repair, the wrong bases sometimes are incorporated into the DNA patch. In this case, the repair process may be repeated to effect the correct pairing. This process is called mismatch repair and it is triggered by nonhydrogen-bonding base pairs. Evidence suggests that the repair process can correct damage resulting from low levels of exposure to genotoxic agents with a high frequency of success. This raises questions about the degree of risk associated with such exposures. The processes of mutation and repair are illustrated in Figure 1.14.
Cell Repair and Regeneration in Toxic Reactions
The ability of a tissue to repair itself after chemical insult is dependent on the percentage of cells damaged, the nature of the damage, and the rate of cell division. Cells of the epithelium (skin, hair, nails) and mucous membranes (gastrointestinal tract, lung, and urogenital tract) are capable of rapid cell division and tissue regeneration. Sloughing of these tissues is protective because it carries away any toxicants accumulated in the cells from low-level exposures. Bone marrow also has a rapid rate of cell division, as do the germ cells of the developing fetus. Damaged bone marrow may regenerate if enough cells survive.
Conversely, such rapidly dividing tissues are vulnerable to destruction when exposed to higher levels of cytotoxic agents. It is for this reason that hair loss, anemia, diarrhea, and gastrointestinal hemorrhage may occur with the use of antineoplastic drugs and following exposure to radiation. These adverse reactions occur because the interval between cell damage and cell division is too short for repair to occur. Cell division cannot proceed or is so impaired that second-generation cells cannot themselves reproduce.
Cells of the liver, kidney, exocrine and endocrine glands, and connective tissue have less rapid rates of cell division but are capable of proliferation and repair.
Heart cells, peripheral nerve cells, and voluntary muscle cells regenerate poorly, but recent evidence suggests that these may be replaced to a greater extent than previously thought. Increased muscle mass from conditioning is due to increased actin and myosin content of the cells.
Fetal Toxicology
Teratogenesis
The existence of malformed infants and animals has been recorded for thousands of years. Early explanations tended to be supernatural. Deformed infants were often regarded as warnings from the gods. The word “monster” comes from the Latin “monstrum,” meaning portent. Malformed animals were thought to be the result of copulation with humans. Those suspected of such an act were often put to death painfully.
Although it was recognized as early as 1932 that dietary deficiencies could result in birth defects, it was not until 1962 that the possibility of drug-induced teratogenesis was recognized as a result of the phocomelia observed in the thalidomide infants in Germany, Japan, the United Kingdom, and other countries including Canada. This tragedy resulted in the introduction of regulatory requirements for tests for teratogenic effects of new drugs in all industrialized countries.
The placenta is a semipermeable membrane that will pass many xenobiotics and their metabolites. Whereas most nutrients cross the placental membrane by energy-consuming, active transport systems, toxicants cross mainly by passive diffusion, so that lipid solubility is an important determinant of toxicity to the fetus. The exceptions are antimetabolites (anticancer drugs), which are analogs of natural substrates and which may utilize their transport pathways. Highly lipid-soluble agents will establish equilibrium between the maternal plasma and the fetus very quickly. More polar agents will take longer to accumulate, but there is no such thing as a “placental barrier.” The placenta possesses biotransforming enzymes so that some detoxication may occur, but it is not enough to protect against any but very low exposures. The human placenta is capable of Phase I and II biotransformations but the enzymes are not inducible. Biological functions in the fetus, even near term, are poorly developed. The blood–brain barrier is imperfect; biotransforming enzymes are undeveloped as is the excretory function of the kidney. The earlier in gestation, the more this is so. The consequences of fetal exposure to toxicants are several. If the exposure is high enough, embryonic death will occur and, possibly, reabsorption of the embryo. This tends to occur early in gestation. If the exposure occurs during structural development, teratogenesis may occur and an anatomical defect may result, the nature of which will reflect the stage of development when the exposure occurred. There is thus a critical period during which a target site exists that does not exist before or afterward. This is what occurred when pregnant women took the drug thalidomide to control morning sickness.
If structural development is more or less complete, a functional defect may occur that may not become evident until later in life when the affected function would normally come into play. The newborn infant may thus be vulnerable to developmental toxicity for several weeks after birth until organ systems become fully matured.
Examples of known teratogenicity resulting from fetal exposure to toxicants include the following (others doubtless exist):
- Folic acid antagonists (aminopterin) used as anticancer agents.
- Androgenic hormones (natural and synthetic progesterones) used to treat breast cancers (and by athletes to increase muscle mass) will masculinize female offspring.
- Thalidomide. A very potent teratogen, use for a single day during day 20–50 of pregnancy was associated with phocomelia (flipper-like limbs). Most animal species are much less sensitive than humans.
- Alcohol. Fetal Alcohol Syndrome (FAS) involves impaired growth, impaired mentation, and distinct facial characteristics (small head, small eye openings, thin upper lip. Fetal Alcohol Effects (FAE) includes only one or two of these criteria.
- Methyl mercury. Infants exposed in utero may develop severe neurological disturbances similar to cerebral palsy. This occurred in the Minamata exposure in Japan.
- Actinomycin D or dactinomycin is an antibiotic that is also used as an antineoplastic agent in certain tumors including Wilms’ tumor and Kaposi’s sarcoma. It is well known to be teratogenic and possibly carcinogenic. It is a potent inhibitor of rapidly dividing cells, both normal and neoplastic. Its potent cytotoxic effect is due to its capacity to bind to DNA in its double helix state. The dactinomycin-DNA complex is very stable so that DNA transcription by RNA polymerase is blocked.
Developmental toxicity can occur in the absence of any maternal toxicity. Teratogenesis may occur from causes other than drugs and environmental chemicals. In fact, in 65%–70% of cases, the cause is never established. Of the remainder, the breakdown is shown in Table 1.4.
Causes of Teratogenesis (Percent of Cases)
Known genetic transmission
11–18
Chromosomal aberration
3–5
Environmental causes
Human-source radiation (therapeutic, nuclear)
<1
Infections (rubella, herpes, toxoplasma, cytomegalovirus, syphilis)
2–3
Maternal metabolic imbalance (endemic, cretinism, diabetes, phenylketonuria, virilizing tumors)
1–2
Drugs and environmental chemicals
2–3
Transplacental Carcinogenesis
The development of cancer as a result of fetal exposure is a possibility if the reproductive system is involved. The best-known example is the occurrence of carcinoma of the vagina and cervix in young women exposed in utero to diethylstilbestrol (DES). DES was given to prevent impending abortion, mostly from 1950 to 1970. Carcinogens abound as natural substances in the environment. Table 1.4 compares some relative risks of these with synthetic ones. The problems associated with attempting to predict the carcinogenic potential of new chemicals from animal studies will be discussed in the next chapter. The problem is critical, due to the high cost and protracted period it takes to evaluate a new chemical.
There is now general awareness that the fetus’ welfare can be threatened by smoking and alcohol consumption by the mother. Avoidance of ingestion, inhalation, or dermal contact of xenobiotics, including recreational drugs, therapeutic agents (where possible) solvents, and other volatile chemicals is to be avoided during pregnancy, especially during the first trimester.
Population and Pollution
It is generally accepted that the root of most anthropogenic environmental problems is overpopulation. This certainly includes chemical carcinogenesis that abounds in our environment (Table 1.4). The increase in the demand for consumer goods, food, and energy resulting from population growth leads to the creation of more waste and pollution. As of April 2012, the world population stood at 7.035 billion. The doubling time of the Earth’s population is down to about 30 years, and this has created incredible pressures on our ability to provide food, living space, energy, and manufactured goods, and particularly on our ability to deal with the waste products of our society (Table 1.5). Our insatiable demand for petroleum products has resulted in drilling in more difficult and hazardous areas such as the arctic where the results of an oil spill would be catastrophic. This subject will be dealt with in more detail later in this text. There has been little official recognition of the population problem in the West, nor specific actions to control population explosion. The People’s Republic of China has long had a restriction of two children per couple in an effort to limit population growth. Controls such as this are repugnant to Western democracies. Western attitudes regarding measures to control population growth are changing, but fear of reaction from religious groups and right-to-life activists, who tend to link population control to issues such as abortion and birth control, has made the issue unattractive to politicians. The lessons of nature should convince us of the need to address this issue. When a population exceeds the ability of its ecosystem to support it, it dies out.
Relative Carcinogenic Hazards in the Environment
Source
Cancers/100,000 Population
Average U.S. lifetime risk of cancer (all types)
25,000–30,000
Foods
Four tablespoons (60 mL) of peanut butter/day (due to aflatoxin)
60
One pint (550 mL)/day (aflatoxin)
14
8 oz (226.8 g) broiled steak/week (nitrosamines, polycyclic aromatic hydrocarbons or PAHs)
3
One diet soda/day (saccharin in the United States, questionable)
70
Average U.S. fish consumption/day
33
Lake Michigan sport fish (based on median consumption levels and EPA potency values)
480–3,300
U.S. sport fish based on (Kim and Stone potency values)
77–340
Drinking water
Average U.S. groundwater based on 2 L/day
1
Niagara River water based on 2 L/day (EPA potency value)
0.3
Air
Various estimates (U.S. urban)
10–560
Table 1.6 lists some carcinogens that may be encountered in the workplace, a striking illustration of how our insatiable demand for consumer goods and energy can put us at risk.
Some Potential Carcinogens Encountered in Industry
1. PAHs (e.g., benzo[a]pyrene); produced by any combustion process, refining and distilling of petroleum, breakdown of lubricants, in welding and foundry processes
2. Benzene (32 million tons/year produced); chemical and petrochemical industries, solvents (paint industry), as an impurity, etc.
3. Carbon tetrachloride (CCl4 ); solvent in industrial processes
4. Halogenated hydrocarbons (PCBs, PBBs, TCDD); in lubricants, transformer insulation, pesticides, etc.
5. Asbestos from mining, in insulation
6. Vinyl chloride in plastics industry
7. Formaldehyde (embalmers, pathologists)
Review Questions
- Match the following terms with the appropriate definition:
- Pharmacodynamic
- Probit
- Economic toxicology
- Xenobiotic
- Anthropogenic
- A substance foreign to living systems
- ii. A chemical process in biological systems in which neither of two agents which interact are permanently altered
- iii. The application of toxicology to achieve an advantage for humankind
- iv. Resulting from human activity
- A probability unit used for making comparisons of potency or toxicity
- Select the correct statement.
- A quantal response is one that is “all or nothing.”
- The response to increasing doses of a drug or toxin continues to increase indefinitely.
- The minimum lethal dose value is applicable to all species.
- A quantal response can never be converted to a graded one.
- Semilogarithmic dose–response plots are used to clean up bad data.
- For Questions 3–10 answer true or false:
- If drug A has a Therapeutic Index (TI) of 500 and drug B has a TI of 10,000, drug A is safer than drug B.
- An acute toxicity reaction is defined as one that occurs within 48 h of exposure.
- Parallel dose–response curves suggest that the two agents in question probably work through the same mechanism.
- If the T1/2 of an agent exceeds the dosage/exposure interval, it will never accumulate in the body.
- Insecticides that inhibit the enzyme acetylcholinesterase cause rapid loss of consciousness.
- Nerve toxins may work by blocking axonal conduction, blocking the enzymatic destruction of a neurotransmitter, blocking the attachment of a neurotransmitter to its receptor, or blocking its release from the nerve terminal.
- Methylmercuries are less toxic than elemental mercury.
- 10. Cadmium is a nontoxic heavy metal.
- For Questions 11–16 use the following code:
- Answer A if statements a, b, and c are correct.
- Answer B if statements a and c are correct.
- Answer C if statements b and d are correct.
- Answer D if only statement d is correct.
- Answer E if all statements (a, b, c, d) are correct.
- 11. a. Most anticancer drugs are teratogenic.
- Genetic abnormalities may involve point mutations or chromosomal breaks.
- Cancer induction is likely a multistage process.
- Viruses have nothing to do with cancer.
- 12. a. Promoters increase tumorigenesis in response to other agents.
- Exposure to the promoter must occur before exposure to the initiator.
- Promoters do not bind to DNA.
- Promoters always cause cancer in their own right.
- 13. a. Selective toxicity is usually absolute, i.e., a substance is completely toxic for the target species and completely harmless for other ones.
- The portal of entry may significantly affect the toxicity and carcinogenicity of a xenobiotic.
- Cancer studies in animals are highly predictable for cancer risk in humans.
- Oncogenes may be activated to convert a cell to a malignant form.
- 14. Highly water-soluble chemicals are
- Excreted by the kidney without biotransformation
- Poorly absorbed from the gastrointestinal tract
- Very polar
- Cross the blood–brain barrier very poorly
- 15. Highly lipid-soluble chemicals
- Are well absorbed from the gastrointestinal tract
- Do not enter the cerebrospinal fluid
- Require biotransformation by the liver before being eliminated by the kidneys
- Generally have a very short T1/2
- 16. Which of the following statements is/are true?
- Serum albumin has numerous binding sites for weakly acidic xenobiotics.
- Substances with epoxide bonds tend to bind irreversibly to macromolecules within cells.
- Mixed-function oxidase enzymes in the SER of the liver cells are responsible for much drug metabolism.
- Conjugation of a xenobiotic with glucuronide is defined as a Phase II reaction.
- For Questions 17–21 answer true or false:
- 17. The kidney is the sole route of elimination of xenobiotics.
- 18. Water-soluble agents of small molecular size pass through the glomerulus.
- 19. Body composition has no influence on the fate of chemicals in the body.
- 20. Some agents may be secreted by an active process across the wall of the renal tubule.
- 21. The presence of one chemical in the body may influence the metabolism of another.
- For Questions 22–24 use the following code:
- Answer A if statements a, b, and c are correct.
- Answer B if statements a and c are correct.
- Answer C if statements b and d are correct.
- Answer D if only statement d is correct.
- Answer E if all statements (a, b, c, d) are correct.
- 22. a. Proto-oncogenes are involved in regulating cell signaling cascades.
- Proto-oncogenes may be converted to oncogenes by retroviruses.
- Oncogenes generally cause uncontrolled cell proliferation.
- Retroviruses may become carriers of oncogenes.
- 23. a. Tumor suppresser genes display dominant characteristics.
- Oncogenes are recessive in nature.
- Tumor suppresser genes have no function in the normal cell.
- Tumor suppresser genes normally prevent uncontrolled cell growth.
- 24. a. “Sense” refers to short segments of DNA coding for some important aspect of cell function.
- “Antisense” is an altered sequence of oligonucleotides matched specifically to a “sense” sequence.
- Antisense sequences inhibit tumor growth in vitro.
- Antisense sequences generally contain 15–25 bases.
- 25. List five chemicals that can induce hepatic microsomal enzymes.
- 26. List five known industrial carcinogens.
- 27. Define the following:
- Teratogenesis
- Carcinogenesis
- Mutagenesis
- 28. List three biological variables that can affect the body’s response to xenobiotics.
- 29. Which of the following statements is/are correct regarding gene therapy for cancer?
- Retroviruses are used as transporters for the therapeutic gene.
- The virus is encapsulated to protect it from the host immune system.
- Preventing expression of oncogenic proteins is a target for some gene therapies.
- None of a, b, or c is correct.
- All of a, b, and c are correct.
- For Questions 30–36 answer true or false:
- 30. D-actinomycin is a teratogenic antibiotic.
- 31. Fetal alcohol effects are a manifestation of teratogenesis.
- 32. There are no natural teratogens.
- 33. All teratogenesis involves anatomical defects.
- 34. The human placenta can biotransform some xenobiotics.
- 35. Most toxicants cross the placenta by active, energy-consuming processes.
- 36. High levels of toxicants are generally more damaging to rapidly dividing tissues.
Answers
- i = d, ii = a, iii = c, iv = e, v = b
- a
- False
- True
- True
- False
- False
- True
- False
- 10. False
- 11. A
- 12. B
- 13. C
- 14. E
- 15. B
- 16. E
- 17. False
- 18. True
- 19. False
- 20. True
- 21. True
- 22. E
- 23. D
- 24. E
- Find the answers in the text for Questions 25–28.
- 29. E
- 30. True
- 31. True
- 32. False
- 33. False
- 34. True
- 35. False
- 36. True
Further Reading
Assennato, G., Cervino, D., Emmett, E.A., Longo, G., and Merlo, F., Follow-up of subjects who developed chloracne following TCDD exposure at Seveso, Am. J. Ind. Med., 16, 119–125, 1989.
Bailey, D.G., Fruit juice inhibition of uptake transport: A new type of food-drug interaction, Br. J. Clin. Pharmacol., 70, 645–655, 2010.
Bailey, D.G., Dresser, G.K., Leake, B.F., and Kim, R.B., Naringin is a major and selective inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice, Clin. Pharmacol. Ther., 81, 495–502, 2007.
Binetti, R., Costamagna, F.M., and Marcelo, J., Exponential growth of new chemicals and evolution of new information relevant to risk control, Ann. 1st Super Sanita, 44, 13–15, 2008.
BRCA1 and BRCA2: Cancer risk and genetic testing, National Cancer Institute Fact Sheet. http://www.cancer.gov/cancertopics/factsheet/Risk/BRCA (accessed September 22, 2011).
Carillo-Infante, C., Abbadessa, G., Bagella, L., and Giordano, L., Viral infections as a cause of cancer, Int. J. Oncol., 30, 1521–1526, 2007.
Cattaneo, R., Miest, T., Shashkova, E.V., and Barry, M.A., Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded, Nat. Rev. Microbiol., 6, 529–540, 2008.
Hardman, J. and Brunton, L.L. (eds.), Blumenthal, D.K., Murri, N., and Hilal-Dandan, R. (assoc. eds.), Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th Edn., McGraw-Hill Medical, New York, 2011.
Hortobagyi, G.N., Toward individualized breast cancer therapy: Translating biological concepts to the bedside, Oncologist, 17, 577–584, April 2, 2012 (Epub ahead of print.)
Klassen, C.D. (ed.), Casarett and Doull’s Toxicology: The Basic Science of Poisons, 7th Edn., McGraw-Hill Medical, New York, 2008.
Klassen, C.D. and Watkins, J.B. III. (eds.), Casarett and Doull’s Essentials of Toxicology, McGraw-Hill Medical, New York, 2010.
Klaunig, J.E., Kamendulis, L.M., and Yong, X., Epigenetic mechanisms of chemical carcinogenesis, Belle News Lett., 9, 2–21, 2000.
Lichtenstein, P., Holm, N.V., Verkasalo, P.K., Iliadou, A., Kaprio, J., Koskenvuo, E., Pukkala, E., Skytthe, A., and Hemminki, K., Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark and Finland, N Eng. J. Med., 343, 78–85, 2000 (comments 135–136).
Marx, J., Research news: Learning how to suppress cancer, Science, 261, 1385–1387, 1993.
McClean, P., Eukaryotic cell cycle and the genetics of cancer. http://www.ndsu.edu./pubweb/∼mcclean/plsc431/cellcycle/cellcycl4.htm (accessed on October 12, 2011).
McPhee, S.J. and Papadakis, M.A. (eds.), Rabow, M.W. (assoc. ed.), Current Medical Diagnosis and Treatment, 51st Edn., Lange Medical Books/McGraw-Hill, New York, 2010.
News and Comment, Animal carcinogen testing challenged, Science, 250, 743–745, 1990.
News and Comment, Experts clash over cancer data, Science, 250, 900–902, 1990.
Oberbauer, R., Not nonsense but antisense—Applications of antisense oligonucleotides in different fields of medicine, Wein. Klin. Wochenschr., 109, 40–46, 1997.
Oncogene, The Broad Institute of MIT and Harvard. http://www.broadinstitute.org/node/1417 (accessed on January 17, 2012).
Putnam, D.A., Antisense strategies and therapeutic applications, Am. J. Health Syst. Pharm., 53, 151–160, 1996.
Research News, Dioxin revisited, Science, 251, 624–626, 1990.
Stephens, P.J., Greenman, C.D., Fu, B., Yang, F., Bignell, G.R., Mudie, L.J., Pleasance, E.D. et al., Massive genome rearrangement acquired in a single catastrophic event during cancer, Cell, 144, 27040, 2011.
Timbrell, J.A., Principles of Biochemical Toxicology, 4th Edn., Informa Health Care, New York, 2009.
Van Loon, J. and Weinshilboum, R.M., Thiopurine methyltransferase isoenzymes in human renal tissue, Drug Metab. Dispos., 18, 632–638, 1990.
Vineis, P., Schatzkin, A., and Potter, J.D., Models of carcinogenesis: An overview, Carcinogenesis, 31, 1703–1709, 2010.
Worldometers: Current world population. http://www.worldometers.info/world-population/ (accessed on November 23, 2011).
Yu, M.-H. (ed.), Environmental Toxicology: Biological and Health Effects of Pollutants, Taylor & Francis Group, Boca Raton, FL, 2011.