


Fumio Hine was born on November 23, 1927 in Osaka Japan. His father was a committed businessman, but Fumio was a bit of a romantic. Nevertheless, after taking some electrical engineering classes in Osaka, he began his studies of chemistry under the direction of Professor Shinzo Okada in the Department of Industrial Chemistry in 1948. Hine received his Ph.D. in 1960 from Kyoto University. He was a Fulbright Scholar, and without much foreknowledge of what to expect, he was sent to work with Earnest Yeager at Case Western Reserve University in Cleveland. During this period he began a close and long collaboration with Robert B. MacMullin. Working in Niagara, MacMullin was well known for his work on the development of the chlor-alkali process and had a great influence on the young Hine. Given that electricity is the major input to most electrolytic processes, such processes are often located close to low-cost electrical power. This was the case in Niagara, New York, which supported a flourishing electrolytic industry beginning in the early 1900s because of Niagara Falls. Cleveland is not too far away, and Fumio recounts visiting MacMullin's home several times in the 1960s, where he discussed concepts of industrial electrochemical processes and explored the natural beauty of the Niagara gorge. In addition to picking up MacMullin's electrochemical engineering philosophy, together they came up with a new phrase, “engineering concepts on industrial processing and electrolyzers.” Probably more than anyone, Fumio developed a view of the electrochemical industry as a chemical process industry, using electrical energy to drive electrochemical reactions.
In 1965, he became a Professor at Kyoto, and in 1969 he moved to the Nagoya Institute of Technology. He is best known for two books: Electrode Processes and Electrochemical Engineering, published in 1985, and Handbook of Chlor-Alkali Technology. The handbook is a comprehensive five-volume set that covers fundamentals, cell design, facility design, and plant commissioning; it was published in 2005. In addition to his academic research, Professor Hine has advised many industries and organizations, particularly on chlor-alkali technology. He has received many awards over his long career, including awards from the Kinki Chemical Society and the Osaka and Japan Soda Industry Association. He was actively involved in and dedicated to the Electrochemical Society, particularly the Industrial and Electrochemical Engineering Division—in 1998 he was named a Fellow of the Society. He has also been active in the American Institute of Chemical Engineering, the Chemical Society of Japan, and the National Association of Corrosion Engineers, where he was a member for more than 50 years.
Professor Hine retired from the Nagoya Institute of Technology in 1991. Fumio Hine, Carl Wagner, Charles Tobias, and Norbert Ibl have been aptly described as the pioneers of Electrochemical Engineering. At the time of the writing of this book, Fumio Hine was living a comfortable life in rural Japan with a large collection of books—about half on electrochemical engineering and half on poetry. Now, Dr. Hine spends much of his time reading and writing Waka poetry. Waka means Japanese poem, and this classical form predates the better known Haiku by more than a thousand years. One of his favorites is Yononaka-ha Nanika Tsunenaru Asukagawa Kinou-no Fuchi-zo Kyou-ha Seni-naru from Kokin Wakashū.
Image Source: Courtesy of Fumio Hine.
Chapter 14
Industrial Electrolysis, Electrochemical Reactors, and Redox-Flow Batteries
The objective of this chapter is to explore a few processes and concepts related to the industrial use of electrochemical systems. In doing so, we will consider mature commercial processes, semicommercial processes, and a process or two that still need development before commercial application is viable. Of course, the economics of these processes will determine their commercial importance. As we examine these industrial processes, we will apply the principles learned in earlier chapters covering thermodynamics, kinetics, and transport.
14.1 Overview of Industrial Electrolysis
The purpose of industrial electrolysis is to use electrical energy to convert raw materials into desired products. This conversion of raw materials takes place in an electrochemical reactor. Since energy is added, the electrochemical cells used are electrolytic rather than galvanic. Hence, the term industrial electrolysis is used to describe these processes. Just two electrolytic processes, the production of aluminum and the chlor-alkali process, consume about 90% of the electricity used in all electrolytic processes. These examples merit special attention in this chapter.
An important difference between industrial electrolytic processes and many of the electrochemical processes that we have considered to this point is the use of flow. Industrial processes frequently operate with a continuous flow of reactants and products. Semicontinuous and batch processes are also used. Reaction rates and, in particular, reaction rates per volume are critical, and can frequently be raised by increasing the transport rate of reactants and products to and from the electrodes. A key parameter in the design of electrochemical reactors is the current density, which determines the area needed and, hence, the number and size of cells required to achieve a desired rate of production. Voltage losses in the cell are also important and determine the energy necessary for production, as well as the energy efficiency of the process. Finally, several important processes involve products in the form of evolved gases.
We begin with an example of an industrial electrolytic process—chlorine production by what is known as the chlor-alkali process. The reactant stream for this process is a purified, saturated brine of NaCl, which flows continuously into the reactor. In addition to chlorine, sodium hydroxide and hydrogen gas are produced simultaneously in the reactor. The two electrochemical reactions are
The standard potential for full cell is 2.188 V. Figure 14.1 illustrates a diaphragm cell, which has been the dominant type of chlor-alkali cell used in the United States for many years. A newer membrane cell is replacing the diaphragm cell in applications across the globe and will be discussed later in the chapter. Early diaphragm cells consisted of vertical anodes of graphite, steel mesh cathodes, and an asbestos separator (diaphragm). Chlorine is evolved at the positive electrode (anode) and the brine flowing in the compartment is called the anolyte. At the cathode, hydrogen is evolved and hydroxide ions are produced. The electrolyte is called the catholyte. A diaphragm keeps the product gases from mixing and is an effective barrier for separating the chlorine and hydrogen. Importantly, however, the diaphragm is permeable to the liquid solution, thus allowing for transport of ions between electrodes. Figure 14.2 shows transport in the diaphragm in more detail. In this design, the solution flows from the anode through the diaphragm to the cathode. This flow helps prevent NaOH from back-diffusing into the anode side of the diaphragm. Within the diaphragm, bulk fluid flow, migration, and molecular diffusion are all present. On the cathode side, the catholyte is removed as one of the products, containing NaOH with some chloride ion contamination. The NaOH is recovered in a subsequent process. Assuming that all of the electrical current goes to support the above reactions, a simple material balance can establish the exit composition if the inlet flow rate and cell current are known. Both Cl2 and H2 are removed from the cell as gaseous products. The composition of NaOH at the exit for this type of cell is ∼12 wt%, and the purity of the chlorine gas produced is 98%. Since chloride ions are consumed at the positive electrode (anode) and hydroxyl ions are produced at the negative electrode (cathode), ions must move to balance the charge; that is, there is an electrical current flow in the solution. Ideally, only Na+ would move from anode to the cathode. This best case is not achieved completely with this design; it is important, nonetheless, to minimize the amount of hydroxide ions that reach the anode and the concentration of Cl− ions that enter the cathode chamber. As you have probably already noted, the diaphragm cell illustrates many important aspects of industrial electrolysis.

Figure 14.1 Diaphragm cell for the production of chlorine and caustic soda.

Figure 14.2 Transport in diaphragm separator of a chlor-alkali cell. (a) Movement of species and potential gradient. (b) Concentration profiles.
About 44 million metric tons of chlorine are produced annually. Assuming that all of this production takes place in diaphragm cells, we estimate the total power required for this production in Illustration 14.1.
We next consider several important performance measures for industrial electrochemical reactors.
14.2 Performance Measures
This section describes three performance measures that are useful for industrial electrolytic systems. We begin with the faradaic efficiency, which is the ratio of the product mass to the amount that could be obtained based on the current and Faraday's law, as was introduced previously in Chapter 1. It can be written as

where mi is the mass of the product, Q is the total charge passed during electrolysis, ![]() is the mass flow rate of the desired product, and I is the total current (assumed constant). In cases where multiple products are formed, the faradaic efficiency for each product may be different. Note that the faradaic efficiency is dimensionless and has the same value if based on mass, moles, or amperes since it is calculated for a single chemical species. In Illustration 14.2, we rework the problem from Illustration 14.1 while accounting for the faradaic efficiency of the chlorine reaction, which for a diaphragm cell is ∼0.96 or 96%. It turns out that the faradaic efficiency for the Cl2 reaction in a chlor-alkali cell is quite high. However, that is not the case for reactions in general, and faradaic efficiencies much less than 1 are frequently encountered.
is the mass flow rate of the desired product, and I is the total current (assumed constant). In cases where multiple products are formed, the faradaic efficiency for each product may be different. Note that the faradaic efficiency is dimensionless and has the same value if based on mass, moles, or amperes since it is calculated for a single chemical species. In Illustration 14.2, we rework the problem from Illustration 14.1 while accounting for the faradaic efficiency of the chlorine reaction, which for a diaphragm cell is ∼0.96 or 96%. It turns out that the faradaic efficiency for the Cl2 reaction in a chlor-alkali cell is quite high. However, that is not the case for reactions in general, and faradaic efficiencies much less than 1 are frequently encountered.
There are a number of reasons why the faradaic efficiency may be less than 1. One of the most important reasons is the presence of side reactions, which are reactions driven by the current flow that do not produce the desired product. We saw previously in Chapter 3 how a current efficiency can be used to account for such reactions. In a chlor-alkali cell, oxygen evolution at the positive electrode is an example of a parasitic (current consuming) side reaction.
The faradaic efficiency is generally different for the anode and the cathode. For example, the evolution of oxygen, an anodic reaction, reduces the amount of chlorine evolved, and thus, lowers the faradaic efficiency of the anode. However, oxygen evolution does not affect the efficiency of the cathode, since it does not influence the amount of caustic or hydrogen produced at that electrode.
Note that the faradaic efficiency as defined in Equation 14.1 is slightly different from the current efficiency introduced in Chapter 3. You should carefully compare the definitions and note the difference. The faradaic efficiency is focused on the final rate of product formation and not just on the electron transfer reaction(s) at the electrode. Therefore, while the current efficiency constitutes an important part of the faradaic efficiency, there are processes that affect the faradaic efficiency, but do not affect the current efficiency. One such process that causes a diminished faradaic efficiency involves the transport of material across the cell. For instance, current is consumed to produce chlorine gas at the positive electrode of a diaphragm cell. This chlorine has a small but significant solubility in the anolyte. The dissolved chlorine can be transported across the diaphragm to the catholyte. In the catholyte, chlorine reacts with NaOH and is not recovered. The chlorine lost to reaction in the catholyte represents a reduction in the faradaic efficiency, even though the current efficiency has not changed. Similarly, hydrogen and caustic can diffuse from the catholyte to the anolyte. This diffusion process lowers the faradaic efficiency and may also contaminate the products.
Contaminants that react with the desired products represent another possible source of lower faradaic efficiency. For example, sodium carbonate in the brine feed reacts with chlorine to reduce the amount of chlorine produced in the process per coulomb passed in the cell,
Finally, product recovery can affect the faradaic efficiency. For example, because of its solubility, a small fraction of the chlorine evolved in a diaphragm cell will be removed with the flow of the anolyte. This chlorine is lost and not recovered as product.
Even though it is not the only contributing factor, the current efficiency remains a critical component of the faradaic efficiency. The current efficiency for multiple electrochemical reactions that take place on a single electrode can be calculated as a function of potential and concentration if the kinetics of the reactions are known as a function of those variables. Such a calculation, useful for process optimization, is given in Illustration 14.2.
The next performance measure of interest is the space–time yield, which is the rate of production per volume of reactor. It is essentially a measure of reactor efficiency and is defined as
where ar, the specific area for the reactor, is the area of the electrode at which the production takes place divided by the volume of the reactor. It is similar to the specific area, a, that was defined in Chapter 5 for porous electrodes. The difference is that the volume used for ar is the total reactor volume rather than just the electrode volume used previously for a. Mi is the molecular weight, and ![]() is the reactor volume. Y has units of kg s−1·m−3. The current, I, and the current density, i, both correspond only to the portion of the current associated with the product of interest. In a situation where there are multiple products, for example, one product at the anode and another at the cathode, a space–time yield can be specified for each product with use of the area of the corresponding electrode. The quantity iar represents the current per unit volume of reactor. Economic analysis is at the heart of industrial processes—both capital and operating costs must be considered. The space–time yield is a parameter that includes the reactor volume, which will directly impact the capital cost. Our main tool to minimize the volume of the reactor is to raise the operating current density. The interplay of the current and volume is apparent from the space–time yield. For a fixed rate of production,
is the reactor volume. Y has units of kg s−1·m−3. The current, I, and the current density, i, both correspond only to the portion of the current associated with the product of interest. In a situation where there are multiple products, for example, one product at the anode and another at the cathode, a space–time yield can be specified for each product with use of the area of the corresponding electrode. The quantity iar represents the current per unit volume of reactor. Economic analysis is at the heart of industrial processes—both capital and operating costs must be considered. The space–time yield is a parameter that includes the reactor volume, which will directly impact the capital cost. Our main tool to minimize the volume of the reactor is to raise the operating current density. The interplay of the current and volume is apparent from the space–time yield. For a fixed rate of production, ![]() , the volume is inversely proportional to the current. Thus, high currents or high current densities lead to smaller volumes and lower capital cost. On the other hand, high current densities result in high cell potentials, and high operating costs. Operating efficiency is addressed below with the third performance measure.
, the volume is inversely proportional to the current. Thus, high currents or high current densities lead to smaller volumes and lower capital cost. On the other hand, high current densities result in high cell potentials, and high operating costs. Operating efficiency is addressed below with the third performance measure.
The magnitude of the current density also influences the type of reactor that can be used economically. When the current density is sufficiently high (>100 A·m−2), then simple two-dimensional electrodes can be used. However, high current densities are not possible for some systems due, for example, to mass-transfer limitations or side reactions. Processes with very low current densities (≤10 A·m−2) may require three-dimensional electrodes, such as the porous electrodes considered in Chapter 5, in order to be economically feasible; such electrodes can have high specific areas of ∼5000 m2·m−3. The reactor configurations considered in this chapter are quite simple. In contrast, a large variety of configurations of varying complexity are used in practice (see Further Reading section for more details).
The third performance measure we consider in this section is the energy efficiency defined as
This parameter is the product of the faradaic efficiency and the ratio of the equilibrium potential to the cell potential. The last term on the right side is the voltage efficiency defined for an electrolytic cell. Physically, the energy efficiency is the theoretical power required to complete the chemical conversion (IRxU) divided by the actual power used (IVcell). A key element of this efficiency is calculation of the cell potential, Vcell, which is discussed in the next section.
14.3 Voltage Losses and the Polarization Curve
In this section, we examine the different polarizations or voltage losses present in typical electrolytic cells. The analysis is largely the same as for any electrochemical cell. Again, the objective is to establish the relationship between the potential of the cell and the current density. Recall from Chapter 4 for an electrolytic cell,
We again use a diaphragm cell as an example. Let's start by examining ohmic losses, which are particularly important for industrial electrolysis cells. The potential drop across a gap of distance h due to current flow between two parallel electrodes is
For the diaphragm cell, however, the situation is a bit more complex as shown in Figure 14.3. Separating the two electrodes is a diaphragm, which is needed to prevent the product gases from mixing. The diaphragm of thickness hd is a porous sheet with an effective conductivity. We can estimate the effective conductivity by modifying the conductivity of the electrolyte to account for the porosity and tortuosity of the diaphragm, similar to what was done in Chapter 5. Additionally, there is a gap between the diaphragm and each of the two electrodes, ha and hc. Finally, there is resistance associated with current flow through and connections to the electrical leads. Thus, the total ohmic resistance of the diaphragm cell is
The first three terms on the right side of Equation 14.5 can be readily evaluated. Rleads depends on the specific connections and bus bars used in the system. For our purposes, you will be given the value for this resistance. Some systems may also have an additional term in Equation 14.5 to account for ohmic losses across imperfect interfaces, frequently referred to as contact resistance.

Figure 14.3 Cell resistances.
Gas evolution occurs in many industrial electrolysis cells and can be associated with a product (e.g., Cl2 in the diaphragm cell) or with side reactions. In Chapter 4, we saw how gas evolution increases the rate of mass transfer in a cell. Gas evolution can also have a negative effect by increasing ohmic losses. As shown in Figure 14.4, gas that evolves at an electrode produces bubbles that rise along the length of the electrode due to buoyancy forces. Gas bubbles displace the electrolyte and cannot carry current. These bubbles reduce the effective conductivity of the solution in a fashion similar to the porous membrane discussed in Chapter 5. One simple expression for this effect, useful for gas fractions up to about 40%, is
where εg is the volume fraction of gas in the gap. As expected, the conductivity decreases as the volume fraction of bubbles increases, providing greater resistance to current flow. A simple way to use this expression is to assume that the distribution of bubbles in the electrolyte is uniform and that the bubbles occupy a certain fraction of the volume, which can be estimated from the height change that occurs in the level of the electrolyte as a result of the gas evolution.

Figure 14.4 Evolution of gas on an electrode.
To more accurately account for the volume fraction of bubbles as a function of height and to connect the local volume fraction explicitly to the local current density, we consider some early work in this area by Charles Tobias that applies at low current densities where the bubble formation is not sufficient to cause circulation of the electrolyte. Specifically, the analysis assumes a stagnant electrolyte, no interaction between bubbles, and a single, constant bubble velocity for all bubbles. The ideal gas law is also assumed to apply. We make the additional assumption that kinetic overpotentials are not significant and that the current is controlled by ohmic losses at these low current densities. With these assumptions, the following expressions result:
where K is the gas effect parameter,
In these equations, ix is the local current density beginning at the bottom of the electrode, where x = 0, and iavg is obtained by integrating the local current density over the electrode surface. The local current density decreases with increasing height due to the presence of more bubbles (see Figure 14.4). Also, as before, h is the gap between electrodes and L is the vertical height of the electrode.
With the assumptions described above, we can also write
Combining Equations 14.8 and 14.10 yields
and
Our goal is to find the potential of the cell that corresponds to a particular current density, which of course is related to the production rate. To do this, we substitute the known current density into iavg in Equation 14.8 and solve for ![]() . Note that K is a function of
. Note that K is a function of ![]() . K also includes the bubble velocity, which depends on the size of the bubble, the difference in density, and viscosity of the liquid. For single, small (less than 0.7 mm in diameter), spherical bubbles at low Re, Stokes flow gives
. K also includes the bubble velocity, which depends on the size of the bubble, the difference in density, and viscosity of the liquid. For single, small (less than 0.7 mm in diameter), spherical bubbles at low Re, Stokes flow gives
Bubble diameters (db) of 0.05−0.1 mm are typical. Use of these equations to calculate ![]() is shown in Illustration 14.5.
is shown in Illustration 14.5.
The procedure just illustrated only applies at very low currents where the electrolyte is stagnant. This condition is satisfied, at least approximately, when the Reynolds number defined earlier in Chapter 4,
is less than about 3; even so, the procedure is frequently used as a first approximation at significantly higher values of Re. Note that the velocity used in Equation 4.48 is the superficial gas velocity defined as
Also, the viscosity and density in Equation 4.48 are based on the liquid properties. Although this method only applies at low current densities, it provides a useful illustration of the effect of bubbles on the ohmic drop. In general, the flow is more complex as bubbles induce electrolyte flow and interact with each other. Some of the factors that are important include coalescence of bubbles, turbulence, the nonspherical shape of the bubbles, and the effect of the walls on the flow. Regardless of the complexity, there is a relationship between the gas evolution rate and the void volume, and that the void volume impacts the ohmic losses in solution.
The impact of gas evolution can be reduced by lowering the current density, increasing the pressure, and increasing the gap distance. At the same time, there are strong incentives to avoid these remedies in order to keep the overall size and cost low, and the energy efficiency high. There is clearly a need for system optimization. One notable engineering solution that has significantly reduced the ohmic resistance associated with bubbles is the use of perforated electrodes in some types of cells that allow gas to be removed from the backside of the electrode out of the current path, specifically the DSA used in the chlor-alkali industry. Another important strategy is the use of convective flow through the gap between electrodes to limit gas buildup by sweeping the gas out of the cell.
In addition to the ohmic losses described previously, kinetic losses or surface overpotentials can also be important. Industrial processes can most often be described with Tafel kinetics, and the corresponding overpotential can be calculated with use of that expression. This is true for a chlor-alkali cell where both electrode reactions are a bit sluggish. For chlorine evolution at the anode,
With ![]() equal to 2, the Tafel slope is roughly 30 mV per decade. Using i0 = 10 A·m−2 at 60 °C, the anode overpotential at a current density of 1940 A·m−2 is about 75 mV. Similarly, for the cathodic hydrogen reduction process,
equal to 2, the Tafel slope is roughly 30 mV per decade. Using i0 = 10 A·m−2 at 60 °C, the anode overpotential at a current density of 1940 A·m−2 is about 75 mV. Similarly, for the cathodic hydrogen reduction process,
With αc = 1 and i0 = 0.07 A·m−2, the overpotential at 1940 A·m−2 is about 0.29 V. The various polarizations or voltage losses at this current density are shown in Figure 14.5. Now that the voltage losses are known, we can estimate the cell voltage with Equation 4.58b to be 3.45 V. Concentration overpotential, which is not likely to be large, has been ignored. For a diaphragm cell operating at 60 °C, the equilibrium potential is 2.25 V. This is the same cell voltage used in Illustration 14.4 to determine the energy efficiency.

Figure 14.5 Polarization in an operating diaphragm chlor-alkali cell.
This methodology can be extended to develop a full polarization curve that relates the cell potential to the operating current density. Just as in other systems, this relationship is essential in designing electrolytic systems.
14.4 Design of Electrochemical Reactors for Industrial Applications
The process of designing an industrial electrochemical system is multifaceted, typically specialized to the application, and iterative. Our description is limited and focuses on the key trade-off between size and efficiency. This balance, illustrated in Figure 14.6, dominates the design process. Economic considerations are at the heart of the design of industrial electrolytic processes. As we'll explore in more detail, low current densities correspond to high efficiencies and low operating costs for electricity. On the other hand, lower current densities require larger electrode areas, and the increased size leads to greater capital costs.

Figure 14.6 Trade-off between size and efficiency is essential part of design.
As noted earlier, there are three important performance measures for electrolytic processes: the faradaic efficiency ![]() , space–time yield (Y), and the energy efficiency
, space–time yield (Y), and the energy efficiency ![]() . These are design variables—the engineer has a hand in selecting these to meet the needs of the application. In contrast, quantities such as exchange-current densities and electrical conductivity are considered physical properties. Additional variables that underlie the performance measures are shown in Table 14.1. A starting point for reactor selection and sizing is the desired production rate of the material,
. These are design variables—the engineer has a hand in selecting these to meet the needs of the application. In contrast, quantities such as exchange-current densities and electrical conductivity are considered physical properties. Additional variables that underlie the performance measures are shown in Table 14.1. A starting point for reactor selection and sizing is the desired production rate of the material, ![]() , typically expressed on an annual basis. The production rate is determined by the opportunity in the market and will include a target price that can be used for profitability analysis. As we know, Faraday's law is used to convert the production rate to a current for use in reactor sizing. For the purposes of this text, the production rate is provided as an input. Detailed costing and profitability analysis is beyond the scope of this text and, if this is your objective, you should refer to a design text that covers economic analyses of processes. Both the current density and cell potential appear in Table 14.1. Of course, the current density and potential are coupled through the polarization curve. This relationship is central to the analysis of electrolytic systems.
, typically expressed on an annual basis. The production rate is determined by the opportunity in the market and will include a target price that can be used for profitability analysis. As we know, Faraday's law is used to convert the production rate to a current for use in reactor sizing. For the purposes of this text, the production rate is provided as an input. Detailed costing and profitability analysis is beyond the scope of this text and, if this is your objective, you should refer to a design text that covers economic analyses of processes. Both the current density and cell potential appear in Table 14.1. Of course, the current density and potential are coupled through the polarization curve. This relationship is central to the analysis of electrolytic systems.
Table 14.1 Eight Key Variables for an Electrochemical Reactor
| Variable | Units | Description | Comments |
| kg·s−1 metric tons·yr−1 | Rate of production of desired material | Size scales with production rate | |
| i | A·m−2 | Current density | These three along with the configuration of the cell would be optimized simultaneously |
| Vcell | V | Potential of an individual cell | |
| ηenergy | – | Energy efficiency | |
| A | m2 | Total electrode area, sometimes referred to as the separator area | Follows directly from Faraday's law once i is established |
| Vs | V | Voltage of the DC power system | |
| m | – | Number of cells that are connected in series | Follows directly, |
| Ac | m2 | Area of individual electrodes | Cells may be placed in parallel |
When designing an industrial electrochemical process, there are many questions to be answered. At what current density should the system operate? Is it preferred to have a few very large cells, or would more, smaller cells be better? What shape should the electrodes take? How should these individual electrodes and cells be arranged? How will flow of reactants be distributed within each cell and between multiple cells? We will discuss these topics in the sections that follow; you should remember, however, that the topics are all interrelated. Before going on, let's look at a quick example where the current density and production rate are given.
Industrial electrolytic cells can be thought of as electrochemical reactors that are used to generate products from raw materials with use of electricity. Some types of reactors can be used for a number of different processes, while others, such as the Hall–Héroult cell used for aluminum production, are tailored for one application. As mentioned previously, a wide variety of reactor types have been developed; in fact, designing electrodes and configuring them to accomplish the chosen reactions efficiently has been an active area of applied and theoretical research. A detailed treatment of a broad spectrum of reactors is beyond the scope of this text. It will be sufficient for us to examine just two basic electrode structures and two means of assembling electrode pairs into an electrochemical reactor. Basic electrode structures of importance here are planar (2D) and porous (3D) electrodes. Configurations will be restricted to (i) assemblies with parallel plate electrodes, which may be 2D or 3D, and (ii) plug-flow reactors with porous electrodes. In the first instance, anode and cathode plates are placed parallel to each other and separated by a fixed distance that is filled with electrolyte as shown in Figure 14.3. When the exchange-current density for the reaction is low, these electrodes are often made porous to increase the specific interfacial area. Almost all the applications that we examine will use these assemblies of parallel plate electrodes. The second approach is limited to porous electrodes or packed beds where reactant streams flow through the electrodes.
Establishment of Operating Current Density
Understanding the relationship between current density and voltage is essential to setting the design current density. There are several factors that influence this choice. One strategy would be to operate at the highest current density possible, which would be at the limiting-current density. As seen from Equation 14.2 for the space–time yield, higher current densities increase Y and result in a smaller reactor volume for a fixed rate of production. A smaller reactor (smaller electrodes and less separator area) corresponds to lower initial costs. However, operation near the limiting current has its drawbacks.
- As the limiting current is approached, Vcell increases rapidly. Both the voltage efficiency and the energy efficiency decrease with increasing current density, see Equation 14.5.
- Operation at the limiting current may lower the current efficiency, especially for multistep reactions. Thus, the faradaic efficiency, ηf, is reduced. A lower faradaic efficiency reduces both the energy efficiency and the space–time yield. This situation is therefore particularly bad—a larger reactor that is less efficient.
- The limiting-current density may exceed the capability of the available electrode materials or separator membranes and may therefore unacceptably shorten the lifetime of these materials. For example, high current operation may lead to excessive cell temperatures that damage the physical components of the cell.
- Constant current operation at the limiting current may not be robust from an operational standpoint, since a change in the inlet conditions or in the operating conditions may reduce the limiting current and lead to undesirable side reactions if operation at the same value of the current is continued.
In the end, the current density will be selected so that the profitability of the electrolytic process is maximized. The choice of current density is both important and complex. Data on reaction rates, mass-transport rates, ohmic losses, current efficiencies, and heat removal are needed for design purposes. For the problems that we will consider in this chapter, you will either be given the current density, instructed to operate at the mass-transfer limit, or be given sufficient information to establish the relationship between the cell voltage and current in order to determine operation below the limiting current. In all cases, we will assume a uniform current density for simplification purposes.
While simplification is necessary for our initial treatment of the topic, let's not forget that we, the electrochemical engineers, have many tools at our disposal to alter the polarization curve. The flow rate of reactants can be increased. Higher flows lead to increased rates of mass transfer and greater limiting-current densities. The gap between electrodes can be reduced, resulting in a lower ohmic drop and better energy efficiency. We may be able to change the concentration of reactants, which directly affects the limiting-current density. Catalysts can be added to reduce kinetic polarizations and improve faradaic efficiency. Porous electrodes may be used to increase the specific surface area of the electrodes, and the temperature of the process may be changed.
Electrical Configurations
Once the current density is fixed, the electrode area follows directly from Faraday's law and the production rate. From our experience with electrochemical systems, we know that the potential of individual cells will be on the order of a few volts, and certainly less than 10 volts. The scale of industrial processes requires enormous amounts of electrical power. Most often the electrical power to drive the electrolytic process comes from high voltage alternating current (AC) that is then rectified. It's not practical to supply vast quantities of direct current (DC) at a potential of just a few volts. The solution is to place cells electrically in series to build voltage. The same approach is used for batteries, double layer capacitors, and fuel cells for high-power applications. The number of series connections is established from the system voltage and potential of an individual cell.
Determining the electrode area and the number of cells connected in series is not the end of the story. When a system contains more than a single anode and cathode pair in series, there are two general methods for making electrical connections: monopolar and bipolar. These terms have the same meanings as they did for batteries and fuel cells. In the monopolar configuration (Figure 14.7a), a separate electrical connection is made to each electrode. The current through the cell is divided among the electrodes electrically connected in parallel, ![]() . Many electrode pairs can be combined in a single cell. In the individual cell, all of the anodes in a cell are at the same potential, as are all of the cathodes. The voltage between each anode and cathode pair is the same and equal to the cell voltage. Both surfaces of each electrode are active. These cells can then be connected in series as needed to add voltage.
. Many electrode pairs can be combined in a single cell. In the individual cell, all of the anodes in a cell are at the same potential, as are all of the cathodes. The voltage between each anode and cathode pair is the same and equal to the cell voltage. Both surfaces of each electrode are active. These cells can then be connected in series as needed to add voltage.


Figure 14.7 Monopolar (a) shown with a separator and bipolar (b) configurations. Current flow is shown for electrolysis. Cell pitch is the number of electrode pairs per unit length.
The second means of connecting multiple electrode pairs is the bipolar stack. Here, assemblies of electrode pairs separated with a solid conductive plate are stacked like a deck of cards. Current in a bipolar stack flows straight through the stack and eliminates the need for connections to each internal electrode; the current distribution tends to be more uniform in the bipolar arrangement. The use of narrow-gap cells is considerably easier in bipolar stacks. There are, however, a couple of important disadvantages. Because the current flows from cell to cell through a bipolar stack, failure of one cell results in failure of the entire stack. In contrast, electrodes in a monopolar arrangement function independently. Also, because the difference in potential from one end of the bipolar stack to the other is large, it is possible for a portion of the current to skip one or more cells and flow directly to another cell downstream (see Figure 14.7b). This phenomenon is referred to as a bypass or shunt current. Bypass currents reduce the faradaic efficiency. Electron-transfer reactions are still needed for the bypass currents, and these reactions may be undesired or destructive to the cell, as is the case for corrosion reactions. Often this damage is more important than the small loss of efficiency. Bypass currents can be reduced by eliminating bypass pathways, but this can be difficult to do in an industrial cell where, for example, electrolyte from different cells flows into a common manifold. Because of these characteristics, bipolar stacks are standard in fuel cells and redox-flow batteries, but used less frequently in industrial electrolysis.
Another key reactor characteristic that needs to be determined is whether or not a divided cell should be used. A divided cell uses a separator to create distinct anolyte and catholyte solutions. If possible, we prefer not to have a separator since it represents an extra resistance to current flow between the electrodes. However, as we have noted previously, the anode is at a higher potential than the cathode in an electrolytic cell. Therefore, a product or by-product produced at the anode can be reduced spontaneously at the cathode. Similarly, a product or by-product produced through reduction at the cathode can be oxidized at the anode. In addition, soluble products may react with each other in solution. Thus, a principal purpose of the separator is to prevent loss of faradaic efficiency by minimizing or eliminating transport of reaction products in order to prevent undesirable reactions. For example, the diaphragm in a chlor-alkali cell helps to keep Cl2 that is dissolved in the electrolyte from reaching the cathode where it would react. Consequently, the faradaic efficiency is increased by preventing Cl2 reduction to Cl− at the cathode. A separator can also maintain purity of the anolyte and catholyte solutions. For example, the diaphragm in a chlor-alkali cell helps to reduce the amount of Cl− in the catholyte, which increases the value of the liquid NaOH product. Finally, separators can prevent the formation of explosive mixtures such as H2/Cl2. The following questions may be useful in considering whether or not to use a separator:
- To what extent is the desired product likely to react at the opposite electrode?
- Are there undesirable solution phase reactions that may be prevented through the use of a separator?
- Are there safety issues that can be addressed through the use of a separator?
- Will use of a separator to create distinct anolyte and catholyte solutions enhance the value of product streams or avoid an expensive downstream separation process?
Flow Configurations
Industrial electrochemical reactors are usually flow reactors. Streams of reactants into and out of the reactor are an essential aspect of both continuous and semicontinuous operation. Reactors can also incorporate internal convection to improve rates of transport, as well as to improve the concentration and temperature distributions. Flow can be important for the removal of gases evolved in the reactor in order to minimize the resistance as discussed in Section 14.3. For assemblies of parallel plate electrodes, the flow is principally coplanar across the electrode surface; whereas with plug-flow reactors, the flow is in void spaces of the porous electrode.
Flow patterns are often unique to the application and too numerous to categorize succinctly. Nonetheless, we will identify the basic flow arrangements for an assembly of cells. For multiple electrode pairs that are housed together in one assembly (a cell for monopolar or a stack for bipolar), there are two principal flow arrangements: parallel flow and series flow. Figure 14.8 illustrates the difference between series and parallel flow for a monopolar design. It is clear that parallel flow will have a lower pressure drop. Series flow enables a greater fraction of the reactants to be converted in a single pass through the assembly, at the expense of a larger pressure drop. Problems associated with gas evolution can be exacerbated with series flow as bubbles accumulate along the flow path. Hybrids of parallel and series are also possible. Industrial practice favors parallel flow where conversion can be increased by placing cell stacks in series with respect to flow, or by separation and recycle of reactants.

Figure 14.8 Basic flow arrangements. (a) Parallel flow. (b) Series flow.
Reactor Volume
The volume of the reactor can be estimated from a knowledge of the electrode area required to meet the desired production rate and the specific area of the reactor, ![]() . Referring back to Figure 14.7, we see that for the parallel plate construction, we can estimate the specific area from the cell dimensions, specifically the thicknesses of the electrodes and the width of the gap between electrodes. The calculation is straightforward, and the result can be summarized by a quantity called cell pitch (see Section 10.4). This parameter is simply the number of electrode pairs per unit length when the repeating units are stacked together.
. Referring back to Figure 14.7, we see that for the parallel plate construction, we can estimate the specific area from the cell dimensions, specifically the thicknesses of the electrodes and the width of the gap between electrodes. The calculation is straightforward, and the result can be summarized by a quantity called cell pitch (see Section 10.4). This parameter is simply the number of electrode pairs per unit length when the repeating units are stacked together.


where the factor of 2 in Equation 14.18b accounts for the two active faces of the electrode. With porous electrodes, there is a lot of internal surface area inside the electrode, represented by a, the specific interfacial area of the electrode. However, even for porous electrodes, we often speak of the superficial current density rather than the true current density based on the internal area. When using the superficial current density with a porous electrode, it is not necessary to include the internal surface area in our analysis.
Scale-Up
The process of designing a system for industrial electrolysis is sequential but with some iteration as noted previously. This characteristic is best illustrated through an envisioned process to scale-up a reactor. Referring to Figure 14.9, we might start with fundamental electrode studies as described in Chapter 6. Basic kinetic data are obtained and side reactions identified. The effects of temperature and reactant concentration are often examined at this stage. The second stage is a complete system of anode, cathode, and electrolyte, but at a subscale—a small single cell where the reactants are supplied in large excess to each electrode. The electrode area of this subscale cell might be a factor of 10 or more less than Ac. Of course, at this point, the area for an individual electrode and the total area are only estimates. These estimates will be refined at each stage. For this subscale cell, uniform current density is assumed and often there are no mass-transfer limitations.

Figure 14.9 One possible scale-up process from fundamental electrochemistry to prototype reactor.
The next step is to increase the area of the cell to its full size; that is, Ac. At this stage, the flow configuration is set and the design includes the effect of finite utilization (conversion) of reactants, u:
Utilization can be defined for an electrode, a cell, or a cell stack. Finally, these individual electrode pairs are connected together to form a system. As noted previously, electrodes are almost invariably connected together electrically (series–parallel combination) to build voltage. The cell may also be combined to form one or more mechanical assemblies. The flow rates of reactants and products are critical elements of the final cell design.
Finally, we note that there is off-the-shelf hardware available for initial evaluation of a process and prototype development. One example is the so-called plate-and-frame assembly shown in Figure 14.10. An off-the-shelf reactor such as this facilitates the development of new electrochemical processes.

Figure 14.10 Plate-and-frame system that is commercially available for process evaluation. Image provided by ElectroCell A/S.
14.5 Examples of Industrial Electrolytic Processes
Industrial electrolytic processes consume about 6% of the total electrical generating capacity of the United States, and represent the principal or only method for producing several important products. This section provides a brief summary of some important industrial applications.
Synthesis of Inorganic Chemicals
Electrolytic production of chlorine and sodium hydroxide, introduced earlier in this chapter, represents the largest electrolytic industry. The process produces chlorine, sodium hydroxide, and hydrogen from a salt solution. Production takes place at 60–95 °C and 0.1–1 MPa. Two types of cells dominated this industry for many years: the diaphragm cell described at the beginning of this chapter and a mercury cell. The mercury cell permitted operation at higher current densities and resulted in products of higher purity relative to the diaphragm cell, with similar energy requirements when the energy required to concentrate the dilute NaOH from the diaphragm cell is included. A combination of new technological developments and environmental concerns has led to the development of a third type of cell, a membrane cell, that takes advantage of a cation-exchange membrane, improved electrodes with reduced overpotentials, and corrosion-resistant polymers for cell construction to produce higher purity products than the diaphragm cell at lower energy consumption rates than the mercury cell. Therefore, most new chlor-alkali cells are of the membrane type.
Although at a much lower production scale, diaphragm and membrane cells are also used industrially to produce chlorine and hydrogen from hydrochloric acid. Hydrochloric acid is a by-product of several important nonelectrochemical industrial processes. Serious environmental concerns associated with handling and disposing of HCl can be avoided by converting it to useful products through electrolysis.
A number of other inorganic chemicals are produced by electrolysis at a smaller scale than chlorine production. For example, fluorine gas and other strong oxidizing agents such as KMnO4, H2O2, and Na2S2O8 (sodium persulfate) can be produced electrochemically. Recently, reagents such as hydrogen peroxide (H2O2) have been produced in situ by electrolysis at the quantity needed for optimal use. Another example of inorganic chemical production by electrolysis is high quality MnO2 for battery applications. In addition to these and other contemporary products, there are many other products that can be made electrochemically, but are not currently manufactured that way because of cost. For example, water electrolysis can be used to produce hydrogen and oxygen gas at high purity; however, except for some specialized applications, other methods of producing hydrogen and oxygen are currently more economical.
Electrowinning of Metals
Electrowinning is the production of metals from ores by electrodeposition from a melt or solution. The most important industrial electrowinning process is the production of aluminum using the Hall–Héroult process. In fact, more electrical power is consumed in aluminum production than in any other electrolytic process. The key innovation, made simultaneously and independently in 1886 by Hall in the United States and Héroult in France, was the discovery that alumina (Al2O3) is soluble in cryolite (sodium hexafluoroaluminate) at about 1000 °C, resulting in a conductive solution. The overall reaction for the production of aluminum is
The precise details of the chemistry are not fully understood; consequently, it is difficult to write a complete set of electrochemical reactions. However, the cathodic reaction is clearly the reduction of aluminum. Molten aluminum is denser than the cryolite solution and falls to the bottom of the crucible where it forms the cell cathode; it is periodically siphoned off as the desired product as shown in Figure 14.11. The carbon anode is consumed in the reaction and is lowered gradually into the cell at a rate of about 2 cm per day to maintain the desired cell gap. The other principal reactant, alumina, is added periodically to the melt through a hopper. Typical faradaic efficiency is near 90%, but energy efficiency is low, on the order of 25%.

Figure 14.11 Hall–Héroult process for the production of aluminum.
Other reactive metals produced by electrowinning from a molten salt include lithium, magnesium, and sodium, where chloride-based salts are typically used.
Copper and zinc are the principal metals recovered by electrowinning from aqueous solutions. The hydrometallurgical process used to do this includes acid leaching followed by extraction and then electrowinning. Historically, most copper has been made by smelting, a competing process. Electrowinning is performed in lined concrete tanks into which alternate rows of anodes and cathodes are placed. The spacing between electrodes is about 5 cm. The operating current density for copper ranges from 150 to 1500 A·m−2, although maximum values of 350–400 A·m−2 are more common. The cathodic reaction is the reduction of the metal, which is plated onto the cathode. The anodic reaction is oxygen evolution on, for example, Pb electrodes. Electrolyte temperatures range from 40 to 60 °C, cell voltages from 1.9 to 2.5 V, and current efficiencies from 80 to 95%. Air sparging, electrolyte circulation, or ultrasonic agitation can be used to increase mass transport and, consequently, the maximum current density. The purity of the copper produced by electrowinning can be quite high (99.999%) and is typically ready for market. In contrast to copper, most zinc is now produced by electrowinning, where the process used is similar to that used for copper. The cell voltage for zinc electrowinning, however, is somewhat higher at 3.3 V.
Electrorefining
In contrast to electrowinning, the purpose of electrorefining is to purify rather than to recover the metal. Aspects of copper electrorefining were used to illustrate several concepts in Chapter 4—you may want to review those parts. Metallic copper, often from a smelting process (approximately 99.5% Cu), is used as the anode. During the refining process, the copper anode is dissolved and copper is plated at the cathode. Any impurities that are more noble than copper stay with the anode and are not dissolved. Impurities that are more active than copper dissolve with the copper into the electrolyte. These active impurities, however, remain in the electrolyte and do not plate out with the copper at the cathode; they are later precipitated out or otherwise removed or recovered from the electrolyte. The net result is the electrodeposition of high-purity copper at the cathode (e.g., 99.999%).
A variety of metals can be purified by electrorefining. For example, nickel, cobalt, lead, and tin can all be refined electrochemically in aqueous solution. Active metals such as aluminum can also be purified in this manner with use of a molten salt electrolyte. The cell voltage for electrorefining tends to be lower than that used for electrowinning as the equilibrium potential is essentially zero for the electrodes of nearly the same composition. Current densities are also modest in order to maintain high purity product and avoid anode passivity (where applicable). As a result, the operating cell voltage for copper electrorefining is only about 0.25 V.
Electrosynthesis of Organic Compounds
A large number and variety of organic reactions can be carried out electrochemically. In fact, organic electrochemistry is considered to be a mature branch of organic synthesis, and most organic reactions that involve electron transfer can be performed by electrochemistry. Types of reactions include oxidation and reduction of functional groups, cleavage, substitutions (e.g., halogenation), additions (e.g., hydrogenation), coupling (e.g., dimerization), and rearrangement. See Further Reading at the end of this chapter for examples of specific reactions.
Organic electrosynthesis reactions may be performed directly or indirectly. Direct synthesis reactions are heterogeneous reactions that take place directly on the surface of the electrode. In most cases, the electrochemical reaction forms a reactive intermediate or radical that undergoes further reaction in close proximity to the electrode surface to produce the desired product. Indirect electrosynthesis reactions take place via a mediator, which in turn reacts homogeneously in solution with the organic reactant to produce the desired product. The mediator is regenerated electrochemically once it has reacted to affect the desired synthesis. Therefore, there are no waste or disposal concerns since the mediator is recycled and not consumed. Most mediators (catalysts) for indirect synthesis are inorganic redox couples such as the following:
- Reductions: Sn4+/Sn2+, Cr3+/Cr2+, Ti4+/Ti3+, Zn2+/Zn, Na+/NaHg
- Oxidations: Ce3+/Ce4+, Cr3+/Cr6+, Mn2+/Mn3+, Mn2+/Mn4+, Ni (OH)2/NiOOH, I−/I2,Br−/Br2,Cl−/ClO−
Indirect reactions may be advantageous when they can be used to replace organic reactions that have a high overpotential and sluggish kinetics or that tend to passivate the electrodes. Indirect reactions are also favored when the redox catalysts can provide enhanced selectivity. The catalyst regeneration and the chemical reaction steps can take place in the same reactor (in-cell) or in different reactors (ex-cell). An ex-cell strategy, made possible through the use of indirect reactions, permits separate optimization of the catalyst and organic reactions. Multiphase reactions are also possible with the catalyst regeneration in the aqueous phase and the organic reaction in a separate organic phase. Use of multiple phases can facilitate product separation and enhance the commercial viability of a process. As always, however, there are trade-offs between the simplicity of a direct process and the enhanced flexibility of an indirect process that must be considered carefully in the design process.
In spite of the many possibilities that exist, relatively few organic electrosynthesis reactions have been successful industrially. Even some of the early successes are no longer performed commercially. Steckhan (2012) estimated that approximately 200 reactions have been performed at the pilot scale with more than 100 commercially available. It is difficult to get a precise number because the details of many of industrial processes are often kept confidential.
The most significant industrial process is the production of adiponitrile, an intermediate in the production of Nylon®. It is the only organic electrosynthesis process where the volume of production is consistent with that of a commodity chemical (300,000 metric tons·yr−1). The reactions are shown below. The cathode reaction is the electro-hydro-dimerization of acrylonitrile, and oxygen is evolved at the anode.
The overall reaction is
These reactions occur in an undivided bipolar stack using aqueous sulfuric acid as the electrolyte. Another process, currently under development, is the electrochemical synthesis of ethylene glycol, which has the potential to become another high-volume process.
There are many potential advantages to the electrochemical synthesis of organic compounds. The inherent advantage is that electrons serve as the oxidizing and reducing agents. These electrons are, in general, inexpensive and clean relative to chemical agents. The rate of reaction is activated with potential rather than temperature. Thus, the mild conditions characteristic of electrochemical synthesis are well suited for chemicals that are heat sensitive. Also, since the current is directly proportional to the reaction rate, these reactions are inherently easier to control. Closely connected to the ability to control the process is the potential for high selectivity from electrochemical processes. Selectivity is particularly important for high-value specialty products. In spite of these advantages, the number of commercial processes is small, as are the volumes produced, as mentioned previously.
Given the advantages of organic electrosynthesis, why are there not more successful industrial processes? What are the key factors that contribute to a successful process? It turns out that energy costs and initial capital costs associated with the electrochemical cells are not typically the problem. At the risk of overgeneralizing, the factors that are often most important are the availability and cost of the reactants, the reaction yield that can be obtained, the ability to inexpensively separate the product(s) from reactant(s), the availability of a suitable, stable electrolyte, and the ability to achieve an acceptable production rate. Because of the low conductivity of organic solvents, it is necessary to add a supporting electrolyte, which must then be separated from the product downstream. Many commercially successful processes involve water soluble reactants and products and utilize sulfuric acid as the electrolyte. Alcohols and acetic acid are also used industrially with some frequency. Cosolvents can be used to enhance solubility if needed. Separation can be facilitated by phase separation where feasible.
Another important factor that may easily be overlooked is the need to consider organic electrosynthesis as a design alternative early in process development. It is difficult and expensive to consider such options at an advanced stage of design. Consequently, it is important for commercial success that a company has the expertise needed to consider electrochemical options as part of their normal design process. This factor is likely to become more important in the future as society shifts to solar power as the primary energy source from which electricity can be generated directly rather than from fuels as is currently the case.
14.6 Thermal Management and Cell Operation
As you may have noted, some of the industrial processes discussed in this chapter operate at high temperatures. The most extreme example considered is the electrowinning of aluminum, which takes place in molten salt at temperatures of almost 1000 °C. How much heat is required to maintain the required temperature and how is this heat supplied? In this section, we consider heating and cooling of electrochemical systems since temperature control is a critical part of any industrial process.
Let's begin with an overall energy balance that applies to a system in which multiple reactions take place. For open systems, the following energy balance applies:

where
 = mass of the system, assumed constant [kg]
= mass of the system, assumed constant [kg] = average heat capacity of the system [J·kg−1·K−1]
= average heat capacity of the system [J·kg−1·K−1] = enthalpy of outlet stream p [J·mol−1]
= enthalpy of outlet stream p [J·mol−1] = enthalpy of inlet stream m [J·mol−1]
= enthalpy of inlet stream m [J·mol−1] = molar flowrate of inlet stream m, [mol·s−1]
= molar flowrate of inlet stream m, [mol·s−1]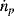 = molar flowrate of outlet stream p, [mol·s−1]
= molar flowrate of outlet stream p, [mol·s−1] = heat transferred to the system from the environment [W]
= heat transferred to the system from the environment [W] = Rate of work done by the system on the environment [W]
= Rate of work done by the system on the environment [W] = Rate of reaction of species i [mol·s−1]
= Rate of reaction of species i [mol·s−1] = Heat of reaction j per mole of species i [J·mol−1]
= Heat of reaction j per mole of species i [J·mol−1]
For the electrolytic systems considered in this chapter, ![]() is positive and equal to the power added to the cell in order to carry out the reaction, IVcell. The heats of reaction apply to full reactions rather than to half-cell reactions, and are determined as described in Chapter 2. When operating at steady state, the term on the left side of Equation 14.22 is zero. Consequently, one important use of the energy balance is to determine the rate of heat (
is positive and equal to the power added to the cell in order to carry out the reaction, IVcell. The heats of reaction apply to full reactions rather than to half-cell reactions, and are determined as described in Chapter 2. When operating at steady state, the term on the left side of Equation 14.22 is zero. Consequently, one important use of the energy balance is to determine the rate of heat (![]() ) that must be added to or removed from the system in order to maintain a steady temperature. Let's illustrate the use of the balance by applying it to aluminum electrowinning (see Illustration 14.12).
) that must be added to or removed from the system in order to maintain a steady temperature. Let's illustrate the use of the balance by applying it to aluminum electrowinning (see Illustration 14.12).
As the illustration demonstrates, heat must be removed from cells used to produce aluminum. This situation is typical for an industrial electrolytic process. Therefore, the focus is on rejecting heat in order to maintain the desired temperature. The high operating temperature for aluminum production facilitates heat transfer from the cell to the environment, which directly provides the necessary cooling. The formation of a solidified molten salt insulating layer on top of the melt permits the system to flexibly maintain the needed temperature. However, these characteristics are not typical of industrial electrolytic cells. For operation closer to room temperature, heat is removed by placing heat exchangers in the anolyte and catholyte loops. In other words, the cooling takes place outside of the reactor with use of heat exchangers placed in the flow loops as illustrated in Figure 14.12. Evaporative cooling towers are frequently used to provide a heat sink for large systems. If operation is assumed to be steady, Equation 14.22 can be used to calculate the heat that would need to be removed to maintain a constant temperature. Similarly, Equation 14.22 can be used to approximate the increase in electrolyte temperature in the electrochemical reactor by assuming adiabatic operation or a known finite heat loss and then calculating the outlet temperature.

Figure 14.12 Process diagram for Zn electrowinning that emphasizes cooling of the electrolyte.
14.7 Electrolytic Processes for a Sustainable Future
Electrolytic Fuel Generation
As we look to the future, it seems clear that the sun will be our primary source of energy. In addition to solar thermal methods, solar energy can be captured in the form of energetic electrons and holes, inherently an electrochemical process. Also, since the availability of solar energy is cyclic, electrochemical processes can be used for energy storage in order to provide the energy needed in off cycles.
Perhaps more important, the transition to solar energy as the primary source of energy for society will likely be accompanied by a shift from fuel-based energy use to direct use of electricity. Electrochemical devices will undoubtedly play a critical role in this shift. Electricity has traditionally been the “high-end” form of energy since it has most frequently been generated using fuels. Consequently, it has historically been more efficient to use fuels directly, where possible, rather than to use fuels to generate electricity, which is subsequently used for the intended application. This will change as electricity is generated directly from renewable sources; hence, electrochemical processes that use electricity directly will have an added economic advantage. There will also likely be a shift from centralized generation of electricity to a distributed solar-based system, which will again impact the use and scale of electrochemical devices. The same type of shift is already occurring in the transportation industry, driven by environmental concerns, where portability will continue to depend on electrochemical devices.
What role, if any, will electrolytic processes play with respect to fuels in a solar-based system? In applications where direct use of electricity is not viable, electric power can be used to generate solar fuels such as hydrogen. This process is a reversal of the previous paradigm where fuels were used to generate electricity. Electrolysis of water to produce hydrogen and oxygen is perhaps the electrolytic process first considered for fuel generation.
Water Electrolysis
The electrolysis of water is essentially a fuel cell in reverse, where electricity is used to create hydrogen and oxygen from water. Therefore, water electrolyzers reflect the types of technologies that we considered for fuel cells in Chapter 9. The three principal types of electrolyzers are alkaline, PEM, and solid oxide.
Most commercial water electrolysis is performed with alkaline electrolyzers. The electrode reactions are as follows:
and
The net reaction is, of course, just the splitting of a water molecule to produce hydrogen and oxygen. Figure 14.13 shows the equilibrium potential and thermoneutral potential (−ΔHRx/nF) for water electrolysis as a function of temperature. These data assume the water is a vapor. Near room temperature, where water is a liquid, the two values are close to one another. At high temperatures, there is large difference—which is important for high-temperature electrolysis. The equilibrium potential, which is proportional to the free energy change for the reaction and, therefore, the minimum potential needed to split the water, decreases with increasing temperature. In contrast, the thermoneutral potential does not change significantly with temperature. Operation at a cell voltage greater than the equilibrium potential but less than the thermoneutral potential would lead to net cooling due to the reversible heat term (TΔS) for water electrolysis, and would require the addition of heat to the reactor. Above the thermoneutral potential, the irreversible losses in the cell are sufficient to generate the heat needed for the reaction. Note that, while convenient, analysis with the thermoneutral potential is approximate for an open system such as an electrolyzer since it does not account for the enthalpy of the inlet and outlet streams. Therefore, where possible, use of the full energy balance, Equation 14.22, is preferred.

Figure 14.13 Equilibrium and thermoneutral potentials assuming gaseous water.
Voltage losses as a function of current density in a typical alkaline electrolyzer are shown in Figure 14.14. The overpotentials for both the anodic and cathodic reactions are significant. As expected, ohmic losses become more important at the higher current densities. Since the energy efficiency is a direct function of the operating voltage, a trade-off exists between the absolute amount of hydrogen that can be produced in a given electrolyzer and the energy efficiency at which it can be produced. Commercial electrolyzers typically operate at cell potentials below 2 V and current densities between 1000 and 3000 A·m−2. Note that at 25 °C, Uθ = 1.229 V and the thermoneutral potential is 1.481 V (assuming liquid water). Given the relatively sluggish kinetics at these temperatures, operation below the thermoneutral potential is not practical.

Figure 14.14 Electrolysis of water at 40 °C.
Source: Adapted from Ulleberg 2003.
Both monopolar and bipolar alkaline electrolyzers are available, although the bipolar configuration is more common. The electrolyte consists of 25–30 wt% KOH, which has a relatively high electrical conductivity. The decision to operate at high rather than low pH is based on material's cost and stability. Specifically, corrosion problems are less severe at high pH than under acidic conditions. Cells traditionally operate at temperatures ranging from 65 to 100 °C. Input or makeup water must be relatively pure (κ < 500 μS·m−1) in order to avoid the buildup of impurities in the cell. Alkaline electrolyzer technology is considered to be mature with a life expectancy of up to 15 years. High-temperature alkaline cells have also been developed; these cells operate at temperatures up to 150 °C in order to improve conductivity and reaction kinetics, although water management is an issue at the high temperatures.
Water electrolyzers based on proton exchange membranes (PEM) are also manufactured. These electrolyzers first appeared during the Space Race in the 1960s, and use technology similar to that of PEM fuel cells. The electrode reactions are as follows:
Hydrogen production rates in PEM electrolyzers are low relative to alkaline cells, in spite of the fact that the current densities are higher. The low production rate is due to the smaller electrode surface area (superficial) in the cell. Use of an ion-exchange membrane provides enhanced safety and increased purity due to the low gas permeability of the membrane. PEM electrolyzers operate well at partial load and have good dynamic performance. The major disadvantages of these electrolyzers are higher initial cost due to the membrane and catalysts, shorter lifetimes, and lower hydrogen production capacity.
A promising technology for water electrolysis that is still at the research stage is that of solid oxide electrolyzers (SOEs). Analogous to solid oxide fuel cells, these devices operate at high temperature and utilize a solid, ceramic O2− conducting electrolyte. The reactions are as follows:
Operation at temperatures up to 1000 °C provides some important advantages. Reaction rates are much faster at high temperature, which circumvents the need for expensive catalysts. Also, the equilibrium voltage decreases with increasing temperature—at 1000 °C U = 0.922 V. That means that less electrical power is needed to drive the reaction in the desired direction to produce hydrogen and oxygen. As seen in Figure 14.13, at 1000 °C, the thermoneutral potential is 1.291 V. It is easy to conceive of a condition where the cell potential is below the thermoneutral potential. This means that, if available, high-temperature heat rather than electricity could be used to provide some of the energy needed to carry out the reaction. Consequently, these SOEs are viewed as a candidate for coupling with high-temperature gas-cooled nuclear reactors to provide both the electrical energy and high-temperature heat for optimal operation. SOEs also provide the advantage of fuel flexibility since, for example, they are capable of directly reducing CO2 to CO or a combination of H2O and CO2 to syngas (H2 and CO) if desired. The primary concern with SOEs is the lack of suitable materials to provide the lifetime needed for industrial applications.
In spite of recent and continuing developments, water hydrolysis is still more expensive, in general, than hydrogen generation from hydrocarbon sources. Consequently, only about 4% of hydrogen is currently produced from water. The largest electrolytic hydrogen plants worldwide are located in proximity to hydroelectric generation facilities in order to take advantage of inexpensive off-peak power.
Other Electrolysis Processes
There are several other electrolytic processes that may offer advantages over water electrolysis. Two key advantages include: (1) use of a waste stream to produce the desired fuel and perhaps other valuable products while simultaneously cleaning up the stream and (2) reduction of the voltage, and hence the energy, required for hydrogen production. It may also be possible to design or modify processes that have traditionally treated hydrogen as an undesirable by-product to produce hydrogen as one of the intended products. Two examples of alternative processes for hydrogen generation include the electrolysis of HCl and the electrolysis of NH3.
Waste streams rich in ammonia can be treated by electrolysis to produce hydrogen gas while simultaneously cleaning up the stream. The equilibrium potential for the electrolysis cell is 0.06 V, much lower than that for water electrolysis (1.229 V). Thus, hydrogen production from ammonia waste streams would require much less energy than that required for water electrolysis. In a similar fashion, urea-contaminated wastewater can also be treated by electrolysis (U = 0.37 V). In addition, both of these processes contribute in a positive way to the reduction of the amount of fixed nitrogen, which has increased significantly as a result of human activity. Finally, another electrochemical process for hydrogen production involves the use of a photoelectrochemical cell. This process will be treated separately in Chapter 15.
Wastewater Treatment
Several of the processes just mentioned involve the use of electrochemical reactors in environment friendly ways to produce useful products while simultaneously cleaning up waste streams. Another use of electrochemical technologies is for the cleanup of industrial effluent streams containing dilute concentrations of toxic materials. To illustrate, electrochemical methods can be used to remove toxic metal ions and have been the subject of renewed interest as regulations have been tightened. For many metals, it is no longer feasible to meet the specified limits for effluent discharge with use of conventional hydroxide precipitation methods. Also, the cost of disposing of the precipitation sludge has increased dramatically as such disposal must ensure that ground contamination by leaching out of the metal ions does not occur. In addition, remediation of water pollution caused by low concentrations of pharmaceutical residues is of significant recent interest and can be done electrochemically. In the treatment below we assume the following:
- The concentration of the contaminant is dilute. Therefore, removal of the contaminant does not change the liquid flow rate to any appreciable extent.
- The removal or cleanup of the contaminant is mass-transfer limited.
- The inlet flow rate and concentration are known.
- The outlet target concentration is known (typically set by regulation).
- Operation is continuous and steady state.
- The reactor cross section is constant and variations only occur in one dimension along the length of the reactor.
- Axial diffusion is not significant.
- Excess supporting electrolyte is used to enhance the conductivity and reduce the potential drop in solution.
The situation that we consider here involves a three-dimensional porous electrode. Our objective is to estimate the size of reactor needed to clean up the stream to the desired level. The flow-through and flow-by configurations, as well as the required material and charge balances, were presented in Chapter 5. Key results are repeated here for convenience. The concentration distribution under mass-transfer control is a function of x only:
where vx is the actual velocity of the fluid in the pores in the direction of interest (εvx would be the superficial velocity). Even though the local rate of reaction is controlled by mass transfer, current must still flow in solution between the upstream counter electrode and the three-dimensional electrode of interest. Again, as we saw in Chapter 5, the current in solution is
Finally, there is a potential drop in solution associated with the current flow. We consider the most common situation where σ ≫ κ. Under such conditions, the change in overpotential is equal to the change in the potential in solution across the thickness of the electrode.
where ![]()
You may be wondering why we care about the potential drop and change in overpotential if the system is at limiting current and controlled by mass transfer. To illustrate, consider Figure 14.14, which shows the current density as a function of overpotential. At limiting current, the current does not change with changing overpotential. However, if the overpotential is increased too much, the current will again increase due to side reaction(s). Side reactions should be avoided since they consume power and may have significant additional negative impacts on the process. For the process under consideration, side reactions are avoided by limiting the potential drop in solution, which limits the range of overpotentials in the system. The maximum ![]() depends on the specific chemical system, but is usually between 100–300 mV. Equation 5.62 can be rewritten as
depends on the specific chemical system, but is usually between 100–300 mV. Equation 5.62 can be rewritten as

where the superficial velocity, vs = ![]() /Ac. Equation 5.57 can be used to relate the concentration at the outlet to the thickness L of the electrode as follows:
/Ac. Equation 5.57 can be used to relate the concentration at the outlet to the thickness L of the electrode as follows:
In writing the equation in this form, we recognize that kcm is a function of the velocity through the bed, vx, which will vary with both ![]() and Ac. The following correlation from Wilson and Geankoplis for packed beds at low Re can be used:
and Ac. The following correlation from Wilson and Geankoplis for packed beds at low Re can be used:
This correlation applies to a packed bed of spherical particles: 0.0016 < Re < 55, 168 < Sc < 70,600, and 0.35 < ε < 0.75. Mass-transfer coefficients in electrochemical reactors typically vary between 10−6 and 10−4 m·s−1.
Our goal is to perform a preliminary calculation of the size of the flow-through reactor. In doing so, we note that there are multiple combinations of cross-sectional area and length that will provide the desired outlet concentration. Also, the simple model used here seems to indicate that the length of the porous bed should be as short as possible since a short length would minimize the potential drop and the pressure drop through the bed (Figure 14.15). However, practical issues such as the nonuniformity of current across a large area and the difficulty of fabricating and obtaining uniform flow over a thin bed with a large cross section become important. Where possible, we recommend that a cross-sectional area consistent with a commercially available electrolyzer be chosen in order to avoid the design and fabrication of a custom system. In practice, the final design decisions will be based on an optimal return on investment.

Figure 14.15 Current potential relationship with a side reaction.
The preliminary design calculation can be performed as follows:
- Choose an initial cross-sectional area for the reactor (best if aligns with that of a commercial available reactor).
- From the total flow rate, cross-sectional area, and void fraction, calculate vs and vx.
- Use vx to estimate kc for the bed with use of a correlation or from experimental data.
- Calculate the length L of the bed with use of Equation 14.24.
- Repeat the above steps for several possible values of the cross-sectional area until a value for L that is comparable to that of the height and width of the unit is obtained.
- Use Equation 14.23 to check if the maximum potential drop has been exceeded for the chosen value of L.
- If the absolute value of the potential drop is lower than the specified maximum, the initial sizing of the electrolyzer is complete. If the potential drop is too high, increase the cross-sectional area until the potential drop no longer exceeds the specified maximum.
Illustration 14.14 demonstrates this general procedure.
14.8 Redox-Flow Batteries
The redox-flow battery is a battery in the sense that it is used to store and release energy. However, it operates much like a combination of a fuel cell (discharge) and an electrolyzer (charge). In contrast to typical secondary batteries where the reactants are part of the electrode, the reactants and products in a flow battery are contained within the electrolyte, which circulates through the cells. As we'll see in a moment, this situation allows for decoupling of the power and energy requirements for the system. There are numerous possible redox couples; here we will focus on the vanadium system as an example.
The vanadium redox-flow battery (VRB) employs two vanadium redox couples for the negative (V3+/V2+) and positive electrodes (VO2+/VO2+) as follows:
Figure 14.16 illustrates how the system operates. A solution (anolyte) circulates through the negative electrode. A separate solution circulates through the positive electrode, the catholyte. Both solutions are acidic, and a proton exchange membrane is used to keep the solutions separate. During discharge, V(II) is oxidized to V(III) at the negative electrode. Electrons travel through the external circuit as usual, and the current in solution is carried primarily by protons. One mole of protons moves across the ion-exchange membrane for each mole of vanadium that is oxidized. These protons react with ![]() and electrons from the external circuit to form
and electrons from the external circuit to form ![]() and water according to Equation 14.27.
and water according to Equation 14.27.

Figure 14.16 Vanadium redox-flow battery. Reactions and transport are shown for discharging.
As just mentioned, the negative and positive electrodes are separated by an ion-exchange membrane (see also PEM, Chapter 9) that prevents the mixing of electrolytes and allows for transport of protons between the electrodes. The ideal characteristics of this separator are low permeability to vanadium cations, high proton conductivity, as well as mechanical and chemical stability.
The electrochemical cell is sized to meet the maximum power [W] and voltage requirements. Independently, the energy requirement [W·h] can be accomplished simply by adding a larger storage tank for reactant-containing electrolyte. This decoupling is shown in Illustration 14.15.
The analysis to find the relationship between current density and potential closely follows the developments made in earlier chapters for electrochemical cells. During discharge, the potential of the cell is the equilibrium potential minus the ohmic, kinetic, and concentration polarizations. Example charge and discharge curves are shown in Figure 14.17.

Figure 14.17 Example of polarization curve for the vanadium redox-flow battery at a high state of charge.
There are many ways to operate the system. It is instructive to write the equilibrium potential for the VRB. With activity coefficients neglected, the change in cell potential with reactant and product concentrations is clear.
To minimize the volume of anolyte needed, we would like to convert nearly all of the V(II) to V(III). There are a couple of ways to effect this conversion. First, we could have the anolyte pass through the cells just once. Achieving full conversion would mean that the concentration of our reactant, V(II), would go toward zero at the exit of the cell. At that point, the equilibrium potential would drop and kinetic and mass-transfer polarizations would become very large. The negative effects associated with this approach could be mitigated to some degree by lowering the operating current density, but that would increase the size of the cell. A second option is to recirculate the anolyte as shown in the figure so that the reactants make many passes through the cell. The conversion during each pass is not high, but after many passes a high overall conversion is achieved. In the extreme, let's imagine that we circulated so rapidly that the anolyte was always well mixed. In this instance, the cell polarizations are minimized but the equilibrium cell potential would change during discharge much like a battery does. These cases are explored further in the problems at the end of the chapter.
Just like other industrial electrochemical systems, economic considerations are paramount for redox-flow batteries. As such, the energy efficiency of the system is a critical metric. We can define efficiency as simply the ratio of the electrical energy out and the electrical energy in, sometimes called round-trip efficiency. If side reactions, crossover, and parasitic power are ignored, this can be approximated with the voltage efficiency of the charge and discharge process,

Using the data from Figure 14.17, we can estimate the current density required to achieve a round-trip efficiency of 65% to be about 2400 A·m−2 (1.35/2.09 = 0.65).
Closure
Industrial electrolysis uses electrical energy to create desirable products via electrochemical processes. In this chapter, we considered several industrial processes of economic importance, including the production of chlorine and aluminum. We also applied the fundamental principles learned previously to help us understand the operation of electrolysis cells. Design criteria for electrochemical reactors were discussed and applied, and methods for approximating reactor size were demonstrated. The thermal management of industrial cells was also considered, with the help of an overall energy balance. Finally, we examined several ways in which electrochemical processes can be used to help achieve a sustainable future. As with any commercial product, economic considerations are critical and the cost of electricity often drives the feasibility of large industrial processes.
Further Reading
- Goodridge, F. and Scott, K. (1995) Electrochemical Process Engineering, Plenum, New York.
- Hine, F. (1985) Electrode Processes and Electrochemical Engineering, Plenum Press, New York.
- Muthuvel, M. and Botte, G. (2009) Trends in Ammonia Electrolysis: Modern Aspects of Electrochemistry. Springer, 45, 207–243.
- Newman, J. and Thomas-Alyea, K.E. (2004) Electrochemical Systems, John Wiley & Sons, Inc., Hoboken, NJ.
- Pletcher, D. and Walsh, F.C. (1993) Industrial Electrochemistry, 2nd edn, Springer Science+Business Media, LLC.
- Rajeshwar, K. and Ibanez, J.G. (1997) Environmental Electrochemistry: Fundamentals and Applications in Pollution Sensors and Abatement, Academic Press, San Diego.
- Steckhan, E. (2012) Electrochemistry, 3. Organic Electrochemistry, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH, Weinheim, Germany.
- Tobias, C.W. (1959) Effect of gas evolution on current distribution and ohmic resistance in electrolyzers. J. Electrochem. Soc., 106, 833.
Problems
14.1. What is the minimum energy required to produce a kg of NaOH from a brine of NaCl? Compare this number with typical values reported industrially of 2100–2500 Wh·kg−1. What are some factors that account for the difference? A rule of thumb for the chlor-alkali industry is that two-thirds of the production costs are electrical energy. If the cost of electricity is $0.06/kWh, estimate the cost to produce a kg of Cl2.
14.2. The third and most modern design for the chlor-alkali process uses an ion-exchange membrane instead of the porous diaphragm. The membrane allows cations to permeate through but is an effective barrier for anions and water. How would the cell design change for this approach? Identify possible advantages and disadvantages to the membrane design.
14.3. An alternative chlor-alkali process has been proposed. Rather than evolving hydrogen, the cathode for a membrane cell design is replaced with an oxygen electrode. Write the cathodic reaction at the oxygen electrode. Compare the equilibrium potential and theoretical specific energy for Cl2 production with that for the conventional cell. What are some advantages and challenges with this concept?
14.4. HCl (g) is a waste product in a number of chemical processes. Ideally, the HCl could be converted to Cl2(g) economically. There are several paths to do this conversion including the Deacon process, which is not electrochemical. You are investigating the direct conversion of anhydrous HCl in an electrochemical cell analogous to a proton exchange membrane fuel cell. A proton-conducting membrane serves as the separator and electrolyte. Write the overall and electrode reactions for this process. What is the equilibrium potential?
14.5. Calculate the theoretical specific energy (W·h·kg−1) to produce Al. Use the reaction shown in Section 14.5 at standard conditions. The U.S. Geological Survey provides the commodity price of Al, http://pubs.usgs.gov/sir/2012/5188/. If the energy efficiency of the process is 30%, what is the maximum price of electricity needed to make the process feasible?
14.6. An aluminum electrowinning process operates at 960 °C. At these conditions, the equilibrium potential is 1.22 V. If the cell is operating at 4.19 V with a faradaic efficiency of 92%, what is the energy efficiency of the process?
14.7. How much electrode area is needed to supply the world with Al? Use a production rate of 20 million metric tons per year. Assume the current density is 900 A·m−2 and a faradaic efficiency of 0.97. What is the annual release of carbon dioxide to the atmosphere? For comparison, in the United States, about 1.5 billion metric tons per year of carbon dioxide are emitted by the transportation sector.
14.8. During electrowinning of aluminum, some of the deposited material dissolves back into the melt. If the rate of dissolution is constant (x g·s−1), develop a relationship for the faradaic efficiency of the process as a function of current density, i. Do the experimental data in the table support this model?
| i [A·m−2] | ηf |
| 441 | 32.8 |
| 803 | 59.0 |
| 1190 | 71.9 |
| 5010 | 93.5 |
| 7520 | 94.6 |
| 9990 | 94.4 |
14.9. Use the parameters from Section 14.3 for the chlor-alkali cell to develop a polarization curve. If the total cost (installation and operating) is given by 0.01 Ac + 5 Vcell, at what current density are costs minimized?
14.10. A key advantage of the DSA for the chlor-alkali process is the removal of gas from the electrode gap. Consider an older design where the gas bubbles remain in the gap. Estimate the optimum electrode gap (minimum ohmic loss) for a 1 m long cell operating at 65 °C, 150 kPa, and 2500 A·m−2. If it were desired to keep the ohmic loss to less than 200 mV to achieve high-energy efficiency, at what current density would the cell need to operate? Use 0.5 mm for the bubble diameter.
14.11. A chlor-alkali plant has a capacity of 35,000 metric tons chlorine per year. If electricity costs are 0.06 [$·kWh−1], what are the annual savings and reduction in production costs per ton of chlorine for each mV of reduction in overpotential? Assume a faradaic efficiency of 0.95. The dimensionally stabilized anode (DSA) is able to reduce the overpotential by about 1 V at 10 kA·m−2. Does describing the DSA innovation as “one of the greatest technological breakthrough of the past 50 years of electrochemistry” seem justified?
14.12. For the diaphragm process shown in Figure 14.1, develop equations for material balances around the cell. Show how to relate the composition of the cathode liquor (caustic) to design parameters. Assume a production rate of 20 kg·h−1 chlorine per hour and that the feed is a saturated brine of NaCl. Include parameters for current efficiency, solubility of chlorine in the brine, and back diffusion of OH−.
14.13. A gas-evolving electrode operates with a gap of 4 mm and the height of the electrode is 0.5 m. The two-electron reaction with a gas-phase product takes place in an undivided cell (no separator) at an average current density of 200 A·m−2. The pressure is 150 kPa and the temperature is 35 °C. Use a bubble velocity of 6 mm·s−1. The conductivity of the solution is 5 S·m−1. It is proposed to reduce the gap to 2 mm to reduce ohmic losses. Is this a good idea? Explain your answer and include a sketch of the current distribution for the two cases.
14.14. For the following processes, indicate whether a divided cell is needed and why. Also indicate whether a bipolar configuration is feasible: (a) electrorefining of tin, (b) production of naphthoquinone (Illustration 14.10), (c) redox-flow battery, and (d) production of adiponitrile.
14.15. A monopolar cell for electrowinning of copper is an open tank that contains multiple parallel plate electrode pairs electrically connected in parallel. If the distance from cathode to cathode is 75 mm, the current density is 280 A·m−2, and the faradaic efficiency is 0.84, calculate the space–time yield. Why is there one more anode than cathode in the cell? Discuss any advantages that might result from making the anodes and cathodes slightly different sizes.
14.16. The cell for electrowinning of zinc is made up of 100 electrode pairs in a monopolar arrangement. Each electrode is 1.4 × 0.7 m. Given an electrode gap of 30 mm, and the i = 3000 A·m−2. The rate of flow of electrolyte is 120 m3·h−1, and enters the cell at a concentration of 100 kg Zn2+ m−3. How would you propose to flow electrolyte to the cells. Sketch the current density on the electrode. Why wouldn't bipolar stack be practical for this application?
14.17. You are setting up a tank house for the electrowinning of Cu. The electrical supply is 180 VDC and 40 kA. For a 1 m2 cell operating at 300 A·m−2, the potential of the cell is 2.0 V and the faradaic efficiency is 85%. Using these conditions, how would you configure the tank house? If the equilibrium potential is 1.45 V, what is the energy efficiency of the process? The cathode cycle is 4 days, during this time by how much does the mass of the cathode increase? Assuming that the tank house operates 340 days per year, what is the annual production rate of copper?
14.18. A large tank house for the electrowinning of zinc contains 200 tanks, each containing 100 electrodes with an area of 1.3 m2 (both sides) in a bipolar arrangement. The electrolyte is acidic. If the operating current density is 490 A·m−2, and the cell potential is 3 V, what is the steady-state cooling requirement for the tank house? If the rate of flow of electrolyte through the cells is 4800 m3·h−1, estimate the change in temperature of the electrolyte through the tank house. Use the heat capacity and density of pure water for this calculation.
14.19. An alternative to the carbon anode in the electrowinning of aluminum is the so-called inert anode. The cathode reaction is unchanged, but here oxygen is evolved instead of consumption of carbon. Write the overall reaction for the inert process. Compare the standard potential for the reaction with the reaction from Equation 14.13. The Hall–Héroult process is already notoriously inefficient. What then are the possible advantages of the inert-anode process?
14.20. Consider the wastewater cleanup with porous electrodes shown in Illustration 14.14. If the particle size were increased to 2 mm, to what value would the effective conductivity need to be increased in order to keep the Hg within the limits. Use an area of 0.75 m2.
14.21. Lithium metal is produced using an electrolytic process. The electrolyte is LiCl-KCl eutectic melt at 427 °C. At these operating conditions, the equilibrium potential is 3.6 V. What is the cathodic reaction? The faradaic efficiency for lithium is reported to be 95%, and the energy consumption is 35 kWh·kg−1. At what potential is the cell operating? What is the energy efficiency of the cell?
14.22. The growth in the lithium-ion battery market has raised demand for lithium. Rechargeable batteries typically use lithiated metal oxides, and the precursor is LiOH not lithium metal. Describe a method to produce LiOH by a process similar to that used for caustic soda using an ion-exchange membrane.
14.23. Calculate the power needed to produce 100 million kg·yr−1 of adiponitrile. Assume the faradaic efficiency is 95% and that the cells operate at a potential of 4.6 V.
14.24. A plant produces 20,000 metric tons of adiponitrile per year. If the power requires is 50 MW, calculate the specific energy (kWh·kg−1) and energy efficiency of the process assuming a faradaic efficiency of 90%. Use 3.08 V for the equilibrium potential of the reaction. How might hydrogen evolution be avoided?
14.25. Compare the energy efficiency of the alkaline electrolyzer from Illustration 14.13 with the efficiency that you would achieve if the current density were doubled and the voltage of the cell increased to 2.25 V. Look at energy efficiency and space–time yield.
14.26. Electrolyzers and fuel cells are envisioned to be a part of an energy storage system for the electrical grid. When supply exceeds demand, hydrogen is generated and stored. When demand exceeds supply, this stored hydrogen is used in a fuel cell to generate electricity. Using the efficiency for electrolysis from Illustration 14.13 and assuming a fuel-cell system efficiency of 60%, what is the round-trip efficiency?
14.27. Calculate change in equilibrium potential as a function of SOC for vanadium redox-flow battery. Assume that the junction region is an ion-exchange membrane that only allows transport of water and protons and completely excludes anion and vanadium ions.

14.28. An iron–chromium redox-flow battery energy storage system is to provide 8 MW of power for a period of 3 hours at a minimum of 650 VDC. The two reactions are
- What is the standard potential of the full cell?
- Assume that the potential of the cell can be treated as ohmically limited. Using the equilibrium potential calculated in part (a) and a resistance of 0.05 mΩ·m2, what cell area is required to provide the desired power while maintaining a round-trip efficiency of 80%? Assume that the current density is the same for charging and discharging.
- If it is impractical to have single cells with an area of greater than 1 m2, what series–parallel configuration would you recommend?
- What is the total volume of electrolyte needed if the solubility of reactants is 0.7 M?
14.29. For the system shown in Illustration 14.15, calculate the rate of heat removal required to maintain a constant temperature. Use a utilization of 0.8 for both the anolyte and the catholyte. The heat capacity can be approximated as that of water. Equation 7.20 can be used to estimate the rate of heat generation.