
Chapter 6. Algorithm Chains and Pipelines
For many machine learning algorithms, the particular representation of
the data that you provide is very important, as we discussed in Chapter 4. This starts with scaling the data and combining features by hand and
goes all the way to learning features using unsupervised machine
learning, as we saw in Chapter 3. Consequently, most machine learning
applications require not only the application of a single algorithm, but
the chaining together of many different processing steps and machine
learning models. In this chapter, we will cover how to use the Pipeline
class to simplify the process of building chains of transformations and
models. In particular, we will see how we can combine Pipeline and
GridSearchCV to search over parameters for all processing steps at
once.
As an example of the importance of chaining models, we noticed that we
can greatly improve the performance of a kernel SVM on the cancer
dataset by using the MinMaxScaler for preprocessing. Here’s code for
splitting the data, computing the minimum and maximum, scaling the data, and
training the SVM:
In[1]:
fromsklearn.svmimportSVCfromsklearn.datasetsimportload_breast_cancerfromsklearn.model_selectionimporttrain_test_splitfromsklearn.preprocessingimportMinMaxScaler# load and split the datacancer=load_breast_cancer()X_train,X_test,y_train,y_test=train_test_split(cancer.data,cancer.target,random_state=0)# compute minimum and maximum on the training datascaler=MinMaxScaler().fit(X_train)
In[2]:
# rescale the training dataX_train_scaled=scaler.transform(X_train)svm=SVC()# learn an SVM on the scaled training datasvm.fit(X_train_scaled,y_train)# scale the test data and score the scaled dataX_test_scaled=scaler.transform(X_test)("Test score: {:.2f}".format(svm.score(X_test_scaled,y_test)))
Out[2]:
Test score: 0.95
6.1 Parameter Selection with Preprocessing
Now let’s say we want to find better parameters for SVC using
GridSearchCV, as discussed in Chapter 5. How should we go about doing
this? A naive approach might look like this:
In[3]:
fromsklearn.model_selectionimportGridSearchCV# for illustration purposes only, don't use this code!param_grid={'C':[0.001,0.01,0.1,1,10,100],'gamma':[0.001,0.01,0.1,1,10,100]}grid=GridSearchCV(SVC(),param_grid=param_grid,cv=5)grid.fit(X_train_scaled,y_train)("Best cross-validation accuracy: {:.2f}".format(grid.best_score_))("Best parameters: ",grid.best_params_)("Test set accuracy: {:.2f}".format(grid.score(X_test_scaled,y_test)))
Out[3]:
Best cross-validation accuracy: 0.98
Best parameters: {'gamma': 1, 'C': 1}
Test set accuracy: 0.97
Here, we ran the grid search over the parameters of SVC using the
scaled data. However, there is a subtle catch in what we just did. When
scaling the data, we used all the data in the training set to compute the minimum
and maximum of the data. We then use the scaled training data to run our
grid search using cross-validation. For each split in the
cross-validation, some part of the original training set will be
declared the training part of the split, and some the test part of the
split. The test part is used to measure the performance of a model trained
on the training part when applied to new data. However, we already used the
information contained in the test part of the split, when scaling the
data. Remember that the test part in each split in the cross-validation
is part of the training set, and we used the information from the entire
training set to find the right scaling of the data. This is
fundamentally different from how new data looks to the model. If we
observe new data (say, in form of our test set), this data will not have
been used to scale the training data, and it might have a different
minimum and maximum than the training data. The following example (Figure 6-1) shows
how the data processing during cross-validation and the final evaluation
differ:
In[4]:
mglearn.plots.plot_improper_processing()

Figure 6-1. Data usage when preprocessing outside the cross-validation loop
So, the splits in the cross-validation no longer correctly mirror how new data will look to the modeling process. We already leaked information from these parts of the data into our modeling process. This will lead to overly optimistic results during cross-validation, and possibly the selection of suboptimal parameters.
To get around this problem, the splitting of the dataset during cross-validation should be done before doing any preprocessing. Any process that extracts knowledge from the dataset should only ever be learned from the training portion of the dataset, and therefore be contained inside the cross-validation loop.
To achieve this in scikit-learn with the cross_val_score function and
the GridSearchCV function, we can use the Pipeline class. The
Pipeline class is a class that allows “gluing” together multiple
processing steps into a single scikit-learn estimator. The Pipeline
class itself has fit, predict, and score methods and behaves just
like any other model in scikit-learn. The most common use case of the
Pipeline class is in chaining preprocessing steps (like scaling of the
data) together with a supervised model like a classifier.
6.2 Building Pipelines
Let’s look at how we can use the Pipeline class to express the
workflow for training an SVM after scaling the data with MinMaxScaler (for
now without the grid search). First, we build a pipeline object by
providing it with a list of steps. Each step is a tuple containing a
name (any string of your choosing1) and an instance of an
estimator:
In[5]:
fromsklearn.pipelineimportPipelinepipe=Pipeline([("scaler",MinMaxScaler()),("svm",SVC())])
Here, we created two steps: the first, called "scaler", is an instance of
MinMaxScaler, and the second, called "svm", is an instance of SVC. Now, we can fit
the pipeline, like any other scikit-learn estimator:
In[6]:
pipe.fit(X_train,y_train)
Here, pipe.fit first calls fit on the first step (the scaler), then
transforms the training data using the scaler, and finally fits the SVM
with the scaled data. To evaluate on the test data, we simply call
pipe.score:
In[7]:
("Test score: {:.2f}".format(pipe.score(X_test,y_test)))
Out[7]:
Test score: 0.95
Calling the score method on the pipeline first transforms the test
data using the scaler, and then calls the score method on the SVM
using the scaled test data. As you can see, the result is identical to
the one we got from the code at the beginning of the chapter, when doing the transformations by hand.
Using the pipeline, we reduced the code needed for our “preprocessing + classification” process. The main benefit of using the pipeline,
however, is that we can now use this single estimator in
cross_val_score or GridSearchCV.
6.3 Using Pipelines in Grid Searches
Using a pipeline in a grid search works the same way as using any other
estimator. We define a parameter grid to search over, and construct a
GridSearchCV from the pipeline and the parameter grid. When specifying
the parameter grid, there is a slight change, though. We need to specify
for each parameter which step of the pipeline it belongs to. Both
parameters that we want to adjust, C and gamma, are parameters of
SVC, the second step. We gave this step the name "svm". The syntax
to define a parameter grid for a pipeline is to specify for each
parameter the step name, followed by __ (a double underscore),
followed by the parameter name. To search over the C parameter of SVC we therefore have to use "svm__C" as the key in the parameter
grid dictionary, and similarly for gamma:
In[8]:
param_grid={'svm__C':[0.001,0.01,0.1,1,10,100],'svm__gamma':[0.001,0.01,0.1,1,10,100]}
With this parameter grid we can use GridSearchCV as usual:
In[9]:
grid=GridSearchCV(pipe,param_grid=param_grid,cv=5)grid.fit(X_train,y_train)("Best cross-validation accuracy: {:.2f}".format(grid.best_score_))("Test set score: {:.2f}".format(grid.score(X_test,y_test)))("Best parameters: {}".format(grid.best_params_))
Out[9]:
Best cross-validation accuracy: 0.98
Test set score: 0.97
Best parameters: {'svm__C': 1, 'svm__gamma': 1}
In contrast to the grid search we did before, now for each split in the
cross-validation, the MinMaxScaler is refit with only the training
splits and no information is leaked from the test split into the parameter
search. Compare this (Figure 6-2) with
Figure 6-1 earlier in this chapter:
In[10]:
mglearn.plots.plot_proper_processing()

Figure 6-2. Data usage when preprocessing inside the cross-validation loop with a pipeline
The impact of leaking information in the cross-validation varies depending on the nature of the preprocessing step. Estimating the scale of the data using the test fold usually doesn’t have a terrible impact, while using the test fold in feature extraction and feature selection can lead to substantial differences in outcomes.
6.4 The General Pipeline Interface
The Pipeline class is not restricted to preprocessing and
classification, but can in fact join any number of estimators together.
For example, you could build a pipeline containing feature extraction,
feature selection, scaling, and classification, for a total of four
steps. Similarly, the last step could be regression or clustering instead
of classification.
The only requirement for estimators in a pipeline is that all but the
last step need to have a transform method, so they can produce a new
representation of the data that can be used in the next step.
Internally, during the call to Pipeline.fit, the pipeline calls
fit and then transform on each step in turn,2 with the input given by the output of the transform
method of the previous step. For the last step in the pipeline, just
fit is called.
Brushing over some finer details, this is implemented
as follows. Remember that pipeline.steps is a list of tuples, so
pipeline.steps[0][1] is the first estimator, pipeline.steps[1][1] is
the second estimator, and so on:
In[15]:
deffit(self,X,y):X_transformed=Xforname,estimatorinself.steps[:-1]:# iterate over all but the final step# fit and transform the dataX_transformed=estimator.fit_transform(X_transformed,y)# fit the last stepself.steps[-1][1].fit(X_transformed,y)returnself
When predicting using Pipeline, we similarly transform the data
using all but the last step, and then call predict on the last step:
In[16]:
defpredict(self,X):X_transformed=Xforstepinself.steps[:-1]:# iterate over all but the final step# transform the dataX_transformed=step[1].transform(X_transformed)# predict using the last stepreturnself.steps[-1][1].predict(X_transformed)
The process is illustrated in Figure 6-3 for two transformers, T1 and T2, and a
classifier (called Classifier).

Figure 6-3. Overview of the pipeline training and prediction process
The pipeline is actually even more general than this. There is no
requirement for the last step in a pipeline to have a predict
function, and we could create a pipeline just containing, for example, a
scaler and PCA. Then, because the last step (PCA) has a transform
method, we could call transform on the pipeline to get the output of
PCA.transform applied to the data that was processed by the previous
step. The last step of a pipeline is only required to have a fit
method.
6.4.1 Convenient Pipeline Creation with make_pipeline
Creating a pipeline using the syntax described earlier is sometimes a
bit cumbersome, and we often don’t need user-specified names for each
step. There is a convenience function, make_pipeline, that will create a
pipeline for us and automatically name each step based on its class. The
syntax for make_pipeline is as follows:
In[17]:
fromsklearn.pipelineimportmake_pipeline# standard syntaxpipe_long=Pipeline([("scaler",MinMaxScaler()),("svm",SVC(C=100))])# abbreviated syntaxpipe_short=make_pipeline(MinMaxScaler(),SVC(C=100))
The pipeline objects pipe_long and pipe_short do exactly the same
thing, but pipe_short has steps that were automatically named.
We can see the names of the steps by looking at the steps attribute:
In[18]:
("Pipeline steps:{}".format(pipe_short.steps))
Out[18]:
Pipeline steps:
[('minmaxscaler', MinMaxScaler(copy=True, feature_range=(0, 1))),
('svc', SVC(C=100, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma='auto',
kernel='rbf', max_iter=-1, probability=False,
random_state=None, shrinking=True, tol=0.001,
verbose=False))]
The steps are named minmaxscaler and svc. In general, the step names
are just lowercase versions of the class names. If multiple steps have
the same class, a number is appended:
In[19]:
fromsklearn.preprocessingimportStandardScalerfromsklearn.decompositionimportPCApipe=make_pipeline(StandardScaler(),PCA(n_components=2),StandardScaler())("Pipeline steps:{}".format(pipe.steps))
Out[19]:
Pipeline steps:
[('standardscaler-1', StandardScaler(copy=True, with_mean=True, with_std=True)),
('pca', PCA(copy=True, iterated_power='auto', n_components=2, random_state=None,
svd_solver='auto', tol=0.0, whiten=False)),
('standardscaler-2', StandardScaler(copy=True, with_mean=True, with_std=True))]
As you can see, the first StandardScaler step was named
standardscaler-1 and the second standardscaler-2. However, in
such settings it might be better to use the Pipeline construction with
explicit names, to give more semantic names to each step.
6.4.2 Accessing Step Attributes
Often you will want to inspect attributes of one of the steps of the
pipeline—say, the coefficients of a linear model or the components
extracted by PCA. The easiest way to access the steps in a pipeline is via
the named_steps attribute, which is a dictionary from the step names to
the estimators:
In[20]:
# fit the pipeline defined before to the cancer datasetpipe.fit(cancer.data)# extract the first two principal components from the "pca" stepcomponents=pipe.named_steps["pca"].components_("components.shape: {}".format(components.shape))
Out[20]:
components.shape: (2, 30)
6.4.3 Accessing Attributes in a Pipeline inside GridSearchCV
As we discussed earlier in this chapter, one of the main reasons to use pipelines is for
doing grid searches. A common task is to access some of the steps of a
pipeline inside a grid search. Let’s grid search a LogisticRegression
classifier on the cancer dataset, using Pipeline and
StandardScaler to scale the data before passing it to the
LogisticRegression classifier. First we create a pipeline using the
make_pipeline function:
In[21]:
fromsklearn.linear_modelimportLogisticRegressionpipe=make_pipeline(StandardScaler(),LogisticRegression())
Next, we create a parameter grid. As explained in Chapter 2, the regularization parameter to tune for LogisticRegression is the parameter C. We use a logarithmic grid for this parameter, searching between 0.01 and
100. Because we used the make_pipeline function, the name of the
LogisticRegression step in the pipeline is the lowercased class name,
logisticregression. To tune the parameter C, we therefore have to
specify a parameter grid for logisticregression__C:
In[22]:
param_grid={'logisticregression__C':[0.01,0.1,1,10,100]}
As usual, we split the cancer dataset into training and test sets, and
fit a grid search:
In[23]:
X_train,X_test,y_train,y_test=train_test_split(cancer.data,cancer.target,random_state=4)grid=GridSearchCV(pipe,param_grid,cv=5)grid.fit(X_train,y_train)
So how do we access the coefficients of the best LogisticRegression
model that was found by GridSearchCV? From Chapter 5 we know that the
best model found by GridSearchCV, trained on all the training data, is
stored in grid.best_estimator_:
In[24]:
("Best estimator:{}".format(grid.best_estimator_))
Out[24]:
Best estimator:
Pipeline(memory=None, steps=[
('standardscaler', StandardScaler(copy=True, with_mean=True, with_std=True)),
('logisticregression', LogisticRegression(C=0.1, class_weight=None,
dual=False, fit_intercept=True, intercept_scaling=1, max_iter=100,
multi_class='warn', n_jobs=None, penalty='l2', random_state=None,
solver='warn', tol=0.0001, verbose=0, warm_start=False))])
This best_estimator_ in our case is a pipeline with two steps,
standardscaler and logisticregression. To access the
logisticregression step, we can use the named_steps attribute of the
pipeline, as explained earlier:
In[25]:
("Logistic regression step:{}".format(grid.best_estimator_.named_steps["logisticregression"]))
Out[25]:
Logistic regression step:
LogisticRegression(C=0.1, class_weight=None, dual=False, fit_intercept=True,
intercept_scaling=1, max_iter=100, multi_class='warn',
n_jobs=None, penalty='l2', random_state=None, solver='warn',
tol=0.0001, verbose=0, warm_start=False)
Now that we have the trained LogisticRegression instance, we can
access the coefficients (weights) associated with each input feature:
In[26]:
("Logistic regression coefficients:{}".format(grid.best_estimator_.named_steps["logisticregression"].coef_))
Out[26]:
Logistic regression coefficients: [[-0.389 -0.375 -0.376 -0.396 -0.115 0.017 -0.355 -0.39 -0.058 0.209 -0.495 -0.004 -0.371 -0.383 -0.045 0.198 0.004 -0.049 0.21 0.224 -0.547 -0.525 -0.499 -0.515 -0.393 -0.123 -0.388 -0.417 -0.325 -0.139]]
This might be a somewhat lengthy expression, but often it comes in handy in understanding your models.
6.5 Grid-Searching Preprocessing Steps and Model Parameters
Using pipelines, we can encapsulate all the processing steps in our machine
learning workflow in a single scikit-learn estimator. Another benefit
of doing this is that we can now adjust the parameters of the
preprocessing using the outcome of a supervised task like regression or
classification. In previous chapters, we used polynomial features on the
boston dataset before applying the ridge regressor. Let’s model that
using a pipeline instead. The pipeline contains three steps—scaling the
data, computing polynomial features, and ridge regression:
In[27]:
fromsklearn.datasetsimportload_bostonboston=load_boston()X_train,X_test,y_train,y_test=train_test_split(boston.data,boston.target,random_state=0)fromsklearn.preprocessingimportPolynomialFeaturespipe=make_pipeline(StandardScaler(),PolynomialFeatures(),Ridge())
How do we know which degrees of polynomials to choose, or whether to
choose any polynomials or interactions at all? Ideally we want to select
the degree parameter based on the outcome of the classification. Using
our pipeline, we can search over the degree parameter together with
the parameter alpha of Ridge. To do this, we define a param_grid
that contains both, appropriately prefixed by the step names:
In[28]:
param_grid={'polynomialfeatures__degree':[1,2,3],'ridge__alpha':[0.001,0.01,0.1,1,10,100]}
Now we can run our grid search again:
In[29]:
grid=GridSearchCV(pipe,param_grid=param_grid,cv=5,n_jobs=-1)grid.fit(X_train,y_train)
We can visualize the outcome of the cross-validation using a heat map (Figure 6-4), as we did in Chapter 5:
In[30]:
plt.matshow(grid.cv_results_['mean_test_score'].reshape(3,-1),vmin=0,cmap="viridis")plt.xlabel("ridge__alpha")plt.ylabel("polynomialfeatures__degree")plt.xticks(range(len(param_grid['ridge__alpha'])),param_grid['ridge__alpha'])plt.yticks(range(len(param_grid['polynomialfeatures__degree'])),param_grid['polynomialfeatures__degree'])plt.colorbar()
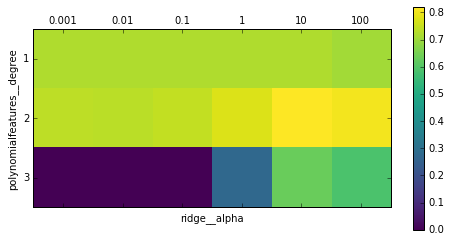
Figure 6-4. Heat map of mean cross-validation score as a function of the degree of the polynomial features and alpha parameter of Ridge
Looking at the results produced by the cross-validation, we can see that using polynomials of degree two helps, but that degree-three polynomials are much worse than either degree one or two. This is reflected in the best parameters that were found:
In[31]:
("Best parameters: {}".format(grid.best_params_))
Out[31]:
Best parameters: {'polynomialfeatures__degree': 2, 'ridge__alpha': 10}
Which lead to the following score:
In[32]:
("Test-set score: {:.2f}".format(grid.score(X_test,y_test)))
Out[32]:
Test-set score: 0.77
Let’s run a grid search without polynomial features for comparison:
In[33]:
param_grid={'ridge__alpha':[0.001,0.01,0.1,1,10,100]}pipe=make_pipeline(StandardScaler(),Ridge())grid=GridSearchCV(pipe,param_grid,cv=5)grid.fit(X_train,y_train)("Score without poly features: {:.2f}".format(grid.score(X_test,y_test)))
Out[33]:
Score without poly features: 0.63
As we would expect looking at the grid search results visualized in Figure 6-4, using no polynomial features leads to decidedly worse results.
Searching over
preprocessing parameters together with model parameters is a very
powerful strategy. However, keep in mind that GridSearchCV tries all
possible combinations of the specified parameters. Therefore, adding
more parameters to your grid exponentially increases the number of
models that need to be built.
6.6 Grid-Searching Which Model To Use
You can even go further in combining GridSearchCV and Pipeline: it
is also possible to search over the actual steps being performed in the
pipeline (say whether to use StandardScaler or MinMaxScaler). This
leads to an even bigger search space and should be considered carefully.
Trying all possible solutions is usually not a viable machine learning
strategy. However, here is an example comparing a
RandomForestClassifier and an SVC on the iris dataset. We know
that the SVC might need the data to be scaled, so we also search over
whether to use StandardScaler or no preprocessing. For the
RandomForestClassifier, we know that no preprocessing is necessary. We
start by defining the pipeline. Here, we explicitly name the steps. We
want two steps, one for the preprocessing and then a classifier. We can
instantiate this using SVC and StandardScaler:
In[34]:
pipe=Pipeline([('preprocessing',StandardScaler()),('classifier',SVC())])
Now we can define the parameter_grid to search over. We want the
classifier to be either RandomForestClassifier or SVC. Because
they have different parameters to tune, and need different
preprocessing, we can make use of the list of search grids we discussed
in “Search over spaces that are not grids”. To assign an estimator to a step, we use the name of the
step as the parameter name. When we wanted to skip a step in the
pipeline (for example, because we don’t need preprocessing for the
RandomForest), we can set that step to None:
In[35]:
fromsklearn.ensembleimportRandomForestClassifierparam_grid=[{'classifier':[SVC()],'preprocessing':[StandardScaler(),None],'classifier__gamma':[0.001,0.01,0.1,1,10,100],'classifier__C':[0.001,0.01,0.1,1,10,100]},{'classifier':[RandomForestClassifier(n_estimators=100)],'preprocessing':[None],'classifier__max_features':[1,2,3]}]
Now we can instantiate and run the grid search as usual, here on the
cancer dataset:
In[36]:
X_train,X_test,y_train,y_test=train_test_split(cancer.data,cancer.target,random_state=0)grid=GridSearchCV(pipe,param_grid,cv=5)grid.fit(X_train,y_train)("Best params:{}".format(grid.best_params_))("Best cross-validation score: {:.2f}".format(grid.best_score_))("Test-set score: {:.2f}".format(grid.score(X_test,y_test)))
Out[36]:
Best params:
{'classifier':
SVC(C=10, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma=0.01, kernel='rbf',
max_iter=-1, probability=False, random_state=None, shrinking=True,
tol=0.001, verbose=False),
'preprocessing':
StandardScaler(copy=True, with_mean=True, with_std=True),
'classifier__C': 10, 'classifier__gamma': 0.01}
Best cross-validation score: 0.99
Test-set score: 0.98
The outcome of the grid search is that SVC with StandardScaler
preprocessing, C=10, and gamma=0.01 gave the best result.
6.6.1 Avoiding Redundant Computation
When performing a large grid-search like the ones described earlier,
the same steps are often used several times. For example, for each
setting of the classifier, the StandardScaler is built again. For the
StandardScaler this might not be a big issue, but if you are using a
more expensive transformation (say, feature extraction with PCA or NMF),
this is a lot of wasted computation. The easiest solution to this
problem is caching computations. This can be done with the memory
parameter of Pipeline, which takes a joblib.Memory object—or just
a path to store the cache. Enabling caching can therefore be as simple
as this:
In[37]:
pipe=Pipeline([('preprocessing',StandardScaler()),('classifier',SVC())],memory="cache_folder")
There are two downsides to this method. The cache is managed by writing
to disk, which requires serialization and actually reading and writing
from disk. This means that using memory will only accelerate
relatively slow transformations. Just scaling the data is likely to be
faster than trying to read the already scaled data from disk. For
expensive transformations, this can still be a big win, though. The other
disadvantage is that using n_jobs can interfere with the caching.
Depending on the execution order of the grid search, in the worst case a
computation could be performed redundantly at the same time by n_jobs
amount of workers before it is cached.
Both of these can be avoided by using a replacement for
GridSearchCV provided by the dask-ml library. dask-ml allows you to avoid redundant
computation while performing parallel computations, even distributed
over a cluster. If you are using expensive pipelines and performing
extensive parameter searches, you should definitely have a look at
dask-ml.
6.7 Summary and Outlook
In this chapter we introduced the Pipeline class, a general-purpose
tool to chain together multiple processing steps in a machine learning
workflow. Real-world applications of machine learning rarely involve an
isolated use of a model, and instead are a sequence of processing steps.
Using pipelines allows us to encapsulate multiple steps into a single
Python object that adheres to the familiar scikit-learn interface of
fit, predict, and transform. In particular when doing model
evaluation using cross-validation and parameter selection using
grid search, using the Pipeline class to capture all the processing steps
is essential for proper evaluation. The Pipeline class also allows
writing more succinct code, and reduces the likelihood of mistakes that
can happen when building processing chains without the pipeline class
(like forgetting to apply all transformers on the test set, or not
applying them in the right order). Choosing the right combination of
feature extraction, preprocessing, and models is somewhat of an art, and often requires some trial and error. However, using pipelines, this
“trying out” of many different processing steps is quite simple. When
experimenting, be careful not to overcomplicate your processes, and
make sure to evaluate whether every component you are including in your
model is necessary.
With this chapter, we have completed our survey of general-purpose tools
and algorithms provided by scikit-learn. You now possess all the
required skills and know the necessary mechanisms to apply machine
learning in practice. In the next chapter, we will dive in more detail
into one particular type of data that is commonly seen in practice, and
that requires some special expertise to handle correctly: text data.