
3
Polycyclic Pigments
Polycyclic pigments consist of anellated aromatic and/or heterocyclic moieties. In commercial pigments, these may range from small systems such as diketopyrrolo-pyrrole derivatives, which feature two five-membered fused rings (DPP pigments), to large systems consisting of eight or nine anellated rings such as flavanthrone, pyranthrone, isoviolanthrone or carbazole-annellated dioxazines. The phthalocyanine skeleton with its polycyclic metal complex is somewhat unique in this respect.
Polycyclic pigments do not contain a hydrazone moiety. As a result, polycyclic pigments are also called ‘non-hydrazone pigments’. Anthraquinone hydrazone pigments are an exception, since they contain a polycyclic anthraquinone moiety as well as a hydrazone group. In this book they are described together with other aminoanthraquinone pigments in chapter 3.7.1.
A series of classical hydrazone pigments (formerly called ‘azo pigments’) contains a small polycyclic moiety such as naphthalene or benzimidazolone. The synthesis and the properties of these pigments are dominated by the hydrazone group. Hence, they are treated as hydrazone (Section 2) and not as polycyclic pigments.
On the other hand there are pigments which contain neither a hydrazone group, nor a polycyclic moiety (except naphthalene), for example triphenylmethane pigments and a series of metal complexes. These pigments will be described in the section ‘Miscellaneous pigments’ (Section 4).
Many polycyclic pigments were used as vat dyes before their development as pigments. In 1934 phthalocyanine derivatives were the first colourants to be launched directly as pigments without having previously been used as dyes. This is one aspect in which the history of the phthalocyanine series differs considerably from that of many vat pigments. The latter became commercially accessible only when synthetic methods were found to improve the quality of vat dyes in such a way that they exhibited adequate pigment properties. The coupling reaction affords hydrazone pigments in the form of very fine powders. Polycyclic pigments, on the other hand, which are frequently prepared in organic solvents, evolve as relatively large crystals (up to 100 µm). Appropriate milling and fine dispersion processes are required (finishing) to obtain a form that is suitable as a pigment.
Most pigments derived from vat dyes are structurally based on anthraquinone derivatives such as indanthrone, flavanthrone, pyranthrone or dibromoanthanthrone. There are other polycyclic pigments that may be used directly in the form in which they are manufactured. These include derivatives of naphthalene and perylene tetracarboxylic acid, dioxazine (Carbazole Violet), and tetrachlorothioindigo. Quinacridone pigments were first introduced in 1958, and in the 1980s DPP pigments were added to the series.
3.1 Phthalocyanine Pigments
The phthalocyanine [1–4] system is structurally derived from the aza-[18]-annulene series, a macrocyclic hetero system consisting of 18 conjugated π-electrons. Two well-known derivatives of this parent structure, which is commonly referred to as porphine, are the iron(III) complex of haemoglobin and the magnesium complex of chlorophyll. Both satisfy the Hückel and Sondheimer (4n + 2)π electron rule and thus form planar aromatic systems.
Copper Phthalocyanine Blue (P.B.15) is the copper(II) complex of tetraazatetrabenzoporphine. As shown below, the mesomeric structures indicate that all of the pyrrole rings simultaneously contribute to the aromatic system:

Scheme 3.1 Molecular structure of P.B.15.
The molecule adopts a planar and completely conjugated structure that exhibits exceptional stability.
Phthalocyanines were initially produced only as pigments. It was not until later that techniques such as sulfonation, chlorosulfonation and chloromethylation made it possible to produce and commercially apply these compounds in dye form.
At present, synthetic routes to more than 40 metal complexes other than the copper complex are known. Apart from a cobalt phthalocyanine pigment (P.B.75), which was introduced to the market at the end of the twentieth century, none of the resulting products, however, has stimulated commercial interest as a pigment. Nickel complexes, though, are found in reactive dyes, while cobalt complexes of this basic structure are employed as developing dyes.
At present, Phthalocyanine Blue and Phthalocyanine Green are amongst the most important organic pigments in the market and are sold in large volume. Also, the printed edition of this book is printed with Copper Phthalocyanine Blue.
As early as 1907, A.V. Braun and J. Tscherniak first obtained phthalocyanine from phthalimide and acetic anhydride [5]. The prepared blue substance, however, was not investigated further. In 1927, de Diesbach and von der Weid, in an attempt to synthesize phthalonitrile from o-dibromobenzene and copper cyanide in pyridine at 200 °C, obtained a blue copper complex. The substance was found to be exceptionally fast to acid, alkali and high temperature [6]. Approximately one year later, in trying to manufacture phthalimide from phthalic anhydride and ammonia, researchers at Scottish Dyes Ltd detected a greenish blue impurity coated onto an apparently defective enamel reactor vessel. (Later analysis showed the product to be the phthalocyanine iron complex.) They repeated the experiments with addition of iron filings or of other metal salts and wrote a patent on the procedure in 1928, stating that ‘the products do not appear to be metal salts of phthalimide’, obviously without knowing that they had obtained phthalocyanines [7].
Coordination complexes of zinc, cobalt and platinum show a stability similar to complexes involving copper and iron. They are stable to concentrated, non-oxidizing acids and bases. The fact that such complexes sublimate at 550–600 °C without decomposing points to their extreme heat stability [8].
In contrast to the ionic complexes of sodium, potassium, calcium, magnesium, barium and cadmium, the ease with which transition metal complexes are formed (high constant of complex formation) can partly be attributed to the suitably sized atomic radii of the corresponding metals. Incorporated into the space provided by the comparatively rigid phthalocyanine ring, these metals fit best. An unfavourable volume ratio between the space within the phthalocyanine ring and the inserted metal, as is the case with the manganese complex, results in a low complex stability.
In 1929, Linsted obtained samples of this complex from ICI chemists (Scottish Dyes Ltd was now owned by ICI). ICI had developed two routes leading to the phthalocyanine iron complex. One method started from phthalic anhydride, iron and ammonia, while the second pathway proceeded from phthalimide, iron sulfide and ammonia. In 1933/34, elucidation of the phthalocyanine structure was credited to Linstead. The corresponding copper and nickel phthalocyanines had been prepared in the meantime. ICI introduced the first Copper Phthalocyanine Blue to the market as early as 1935, and the Ludwigshafen subsidiary of the IG Farbenindustrie followed suit with a corresponding product.
In Germany, Phthalocyanine Green was first prepared commercially in 1938, followed by US companies in 1940.
Most phthalocyanine green pigments are derived from copper polychlorophthalocyanines. Chlorobromo derivatives provide a yellowish green shade.
Copper Phthalocyanine Blue exhibits 11 crystal modifications. The metal-free ligand has only five known polymorphs. Its greenish blue crystal phase was used on a large industrial scale for a certain period of time. Free-base Phthalocyanine Blue was largely displaced by β-Copper Phthalocyanine Blue as it became possible to produce the latter more economically (Section 3.1.2.3).
3.1.1 Starting Materials
Heading the list of suitable organic starting materials [9] for phthalocyanine pigments are phthalic anhydride and phthalonitrile.
3.1.1.1 Phthalic Anhydride
Phthalic anhydride is prepared by oxidizing o-xylene. The oxidation may be performed either in the gas phase with vanadium pentoxide as a catalyst or in the liquid phase with dissolved manganese, molybdenum or cobalt salts as catalysts:

At the present time, gas-phase oxidation is the favoured technique.
A third option involves oxidizing naphthalene, possibly with vanadium pentoxide as a catalyst.
Phthalic anhydride is a significant commercial product. Its main area of application is in synthetic resins and plasticizers. In 2014, the production volume worldwide was about 4.5 million tons.
3.1.1.2 Phthalonitrile
Phthalonitrile (also called ‘phthalodinitrile’) is obtained by oxidizing o-xylene with ammonia. To this end, o-xylene is treated with ammonia, either in the presence of an oxidation catalyst or with the aid of oxygen and a catalyst, at 330–340 °C:
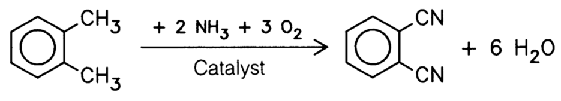
3.1.2 Manufacture
In 1937, Du Pont started producing Copper Phthalocyanine Blue in the USA after it had previously been launched in Great Britain and Germany. Other companies followed suit.
Copper Phthalocyanine Blue was no longer open for patent application after it had been described by de Diesbach and von der Weid in 1927. Yet a great many synthetic methods for this important compound were patented.
Metal phthalocyanine complexes may be obtained by one of the following methods. They can be prepared from:
- phthalonitrile or substituted phthalonitriles and metals and/or metallic salts,
- phthalic anhydride, phthalic acid, phthalic esters, diammonium phthalate, phthalamide or phthalimide and urea, metal salt and a catalyst,
- o-cyanobenzamide and a metal or a metallic salt.
Only two techniques have gained commercial importance. The phthalonitrile process, developed in England and Germany, is particularly important in Germany, while the phthalic anhydride/urea process has stimulated more interest in Great Britain, the USA and Asia.
The technical importance of the phthalonitrile process is only second to the phthalic anhydride/urea technique, of which two varieties are commercially used (Section 3.1.2.2). BASF is the primary European user of the phthalonitrile method.
Knowledge of the most important types of copper phthalocyanine pigments is useful for the understanding of the processes concepts underlying pigment manufacture. Heading the list are the α- and β-modifications of unsubstituted Copper Phthalocyanine Blue (Section 3.1.2.3). The α-modification is sold as an unstabilized product, and a product stabilized against change of crystal modification.
Likewise, halogenated copper phthalocyanine green pigments also have a major impact on the market (Section 3.1.2.5).
3.1.2.1 Phthalonitrile Process
The first technical process involved heating phthalonitrile with copper bronze or copper(I)chloride at 200–240 °C in copper pans. Several variations of this technique were developed in Germany prior to the Second World War. The reaction was performed either without or in the presence of a solvent. A basic distinction is commonly made between the baking process and the solvent process; both may be carried out either by continuous or by batch technique.
3.1.2.1.1 Baking Process
Baking the starting materials in the absence of solvents involves heating phthalonitrile with copper(I)chloride partially in a nitrogen stream to 140–200 °C. Copper(I)chloride is sometimes replaced by copper powder, copper(II) chloride, or pyridine/CuCl2 complex. Baking was originally performed as a batch process on plates which were heated indirectly with high pressure steam. Continuous baking, on the other hand, involved using an electrically heated moving copper belt, a tunnel kiln or a tunnel drying oven.
A considerably exothermic reaction is initiated at a starting temperature of 140–160 °C, and more than 300 °C may be reached during conversion. Partial cleavage under such conditions affects not only the yield but also the quality of the product. Improved temperature control is achieved by either working by continuous operation (see below) or by adding inert inorganic salts, such as sodium sulfate or sodium chloride. It is also possible to cool the reaction mixture during the appropriate phase of the process.
Baking is currently performed by continuous operation. Modern variations involve using heated crushing or milling equipment, such as kneader dispersers or oscillating mills at approximately 200 °C [10]. This technique significantly improves the reaction control over a batch process. If baking is performed by continuous process, phthalonitrile only remains within the reaction vessel for a very short period of time (3–20 min). It is important to remember that the temperature may not exceed 250 °C. The product that evolves from this process is usually purified by acid treatment.
The yield may range from 70% to 80%, although some continuous techniques have claimed as much as 85%. In view of the fact that reaction control is relatively difficult during baking and that the process only affords a limited yield, it is the solvent method that has stimulated particular interest, especially as improved industrial processes were developed. Other trends, however, show a reverse tendency: the baking process is regaining importance. This is mainly on the grounds of economical, ecological and physiological concerns.
3.1.2.1.2 Solvent Process
The solvent process involves treating phthalonitrile with any one of several copper salts in the presence of a solvent at 120–220 °C [11]. Copper(I)chloride is most important. The list of suitable solvents is headed by those with a boiling point above 180 °C, such as trichlorobenzene, nitrobenzene, naphthalene and kerosene. A metallic catalyst such as molybdenum oxide or ammonium molybdate may be added to enhance the yield, to shorten the reaction time and to reduce the necessary temperature. Other suitable catalysts are carbonyl compounds of molybdenum, titanium or iron. The process may be accelerated by adding ammonia, urea or tertiary organic bases such as pyridine or quinoline. As a result of improved temperature maintenance and better reaction control, the solvent method affords yields of 95% and more, even on a commercial scale. There is a certain disadvantage to the fact that the solvent reaction requires considerably more time than dry methods.
After completion of the reaction, the solvent is removed by filtration. Most often, however, the solvent is separated from the crude Copper Phthalocyanine Blue by distillation.
The solvent method may also be performed either by continuous (in cascades) or by batch operation. Continuous techniques in particular have gained considerable technical importance. A phthalonitrile/copper chloride solution is typically treated at 120–140 °C in a flow tube furnace and the temperature subsequently increased to 180–250 °C. The entire process requires approximately 1.5–2 h and affords the pigment in practically quantitative yield. The excellent purity of the product eliminates the need for additional purification with dilute acid or base prior to finishing, a procedure that plays a major role in the baking process. These are the advantages that make the baking process economically favourable, despite the problems connected with the separation and regeneration of the solvents.
The phthalonitrile process has the particular advantage over the phthalic anhydride process of forming ring-substituted chloro-copper phthalocyanines. Using copper(I)chloride produces so-called ‘semi-chloro’ Copper Phalocyanine Blue, a pigment that possesses a statistical average of 0.5 chlorine atoms per copper phthalocyanine molecule. Copper(II)chloride, on the other hand, affords a product that consists of an average of one chlorine atom per copper phthalocyanine molecule. A prerequisite for the formation of the chloro substituted compound, however, is the absence of ammonia or urea in the reaction mixture.
This simple one-step route leads to the starting material for the solvent-stabilized α-modification of Copper Phthalocyanine Blue.
The synthesis of the chlorine-free crude pigment by either the continuous or the batch version of the phthalonitrile process involves adding up to 20% urea or ammonia to the reaction mixture. The latter will provide an effective chlorine trap.
Moreover, the phthalonitrile process has the added advantage of being the more elegant of the two syntheses. This technique makes it possible to produce comparatively pure copper phthalocyanine without obtaining substantial amounts of side products, a phenomenon that is understandable in view of the fact that the phthalonitrile molecule provides the parent structure of the phthalocyanine ring. Formally, rearrangement of the bonds necessitates donation of two electrons to the system:

The advantages of the phthalonitrile process are that phthalonitrile is not only much more costly than phthalic anhydride but also less easily available. In view of the intermediates found so far, and in conjunction with a study of the thermal course of the reaction using differential scanning calorimetry, a reaction mechanism has been proposed for the phthalonitrile route that may be visualized as follows [12].
The reaction is initiated by attack of a nucleophile (Y−), usually the counterion associated with the Cu2+ ion, at one of the CN groups of the phthalonitrile, which is activated by its coordination with the Cu2+ ion. Subsequently, a cyclization reaction to an isoindoline derivative takes place. These steps are repeated three times by a series of similar reactions, finally resulting in a cyclization to a CuPc ring intermediate, whose formation is facilitated by the coordinating role of the copper ion. With a copper(II) salt as reactant it is suggested that Y+ is eliminated from the intermediate, for example the Cl+ ion in the case of CuCl2. The monochloro derivate of CuPc is than formed by electrophilic attack of Cl+ on the CuPc initially formed:

3.1.2.2 Phthalic Anhydride/Urea Process
Likewise, this process [13] may also be carried out either as a solvent-free (baking) method or in the presence of solvents. Although initially performed as a solvent-free technique, it is the solvent version that currently dominates the field of copper phthalocyanine production from phthalic anhydride and urea. However, this trend is being reversed, such that solvent-free methods are attracting interest, especially for ecological reasons.
3.1.2.2.1 Baking Process
The first commercial copper phthalocyanine synthesis, a baking process, involved melting phthalic anhydride with urea at 150 °C in the presence of boric acid. Copper(I)chloride was then added and the temperature increased to approximately 200 °C until the copper phthalocyanine production was completed. The reaction mixture was cooled and the crude product milled. After being washed, first with dilute sodium hydroxide solution and then with dilute sulfuric acid, the material was filtered off and dried. The crude copper phthalocyanine obtained was then dissolved in sulfuric acid and precipitated in ice water (‘acid pasting’) to afford a form that could be used as a pigment. In subsequent years, boric acid in its role as a catalyst largely gave way to various metallic salts. Best results were achieved with molybdenum trioxide and primarily with ammonium molybdate, both of which are currently used. It thus became possible to increase the yield from approximately 50% to more than 90% of the theoretical maximum. Copper(II) chloride may also be replaced by copper(I) chloride, copper carbonate or, most commonly, copper sulfate.
The baking process has remained much the same until the present day; at a stoichiometric ratio of 1 : 4, phthalic anhydride or phthalic acid reacts with an ammonia-releasing compound. The reaction may also start from other suitable materials, such as phthalic acid derivatives, including phthalic acid esters, phthalic acid diamide or phthalimide. Appropriate ammonia releasing agents include urea and its derivatives, such as biuret, guanidine and dicyanodiamide. The fact that a certain amount of urea decomposes to form side products makes it necessary to use excess urea. Approximately 0.2–0.5, preferably 0.25, equivalents of copper salt should be added for each mole of phthalic anhydride; 0.1–0.4 moles of molybdenum salt per mole of phthalic anhydride is sufficient. The reaction temperature is between 200 and 300 °C.
The baking process, particularly the batch variety, presents several serious disadvantages. Not only does the reaction produce solid urea decomposition products, but it also releases large amounts of ammonia and ammonium salts, which escape by sublimation. The foam that is thus formed makes for a porous reaction mixture, which in turn even prevents heat conduction. Moreover, the reaction mixture tends to adhere to the surface of the reaction vessel and the stirring unit, a phenomenon that adds to the complexity of the problem.
At least some of these disadvantages may be overcome by vigorous stirring or milling during the reaction. A baking by batch process has been described that involves heating the reaction components in layers on metal sheets. However, such processes require precise control. If a copper phthalocyanine complex is prepared by batch process in a ball or a pin-type mill, the reaction mixture must be allowed to cool before it is discharged. Reaction times of originally 5–45 min thus create cycles lasting up to 3 h, a highly uneconomical duration.
Continuous baking processes may be conducted, for instance, by treating the reaction mixture in a heated cylinder with a screw drive. The desired thin film, however, can only be produced in large elaborate reaction units, a requirement that makes its manufacture expensive. The same is true for performing the baking process in heated rotating drums. A certain amount of product (namely, the amount that is produced in 2 h) must already be present as the starting materials are added in order to prevent the materials from sticking to the unit. Long reaction times and unprofitable filling levels produce a space–time yield that is unattractive for industrial purposes.
On the other hand, economically advantageous routes by continuous baking process have also been described. The processes are carried out in self-cleaning mixing apparatus such as double screw extruders. More recent patent literature claims yields as high as 80%.
Invariably, the primary product is a crude Copper Phthalocyanine Blue with insufficient properties. It is boiled with dilute hydrochloric acid or aqueous alkali and then rinsed with hot water to remove acid or base before it can be finished.
3.1.2.2.2 Solvent Process
There is somewhat less of a demand on the reaction equipment if phthalic anhydride and urea react in an organic solvent. In principle, the presence of a solvent makes almost no difference to the synthesis by baking process. Using suitable inert high-boiling solvents such as kerosene, trichlorobenzene or nitrobenzene obviates many of the difficulties connected with the reaction mixtures obtained in the baking process. Solvents make it much easier to adequately mix the components and to ensure even heat transfer. Long reaction times, however, remain somewhat of a problem, although suitable temperature control during condensation makes it possible to produce the complex in almost quantitative yield.
The solvent is removed from the solid product by filtration or centrifugation to afford a crude Copper Phthalocyanine Blue of a quality that makes intermediate purification unnecessary. In contrast to the product obtained by the baking process, this material is pure enough to be used directly for further pigment manufacture. Crude Copper Phthalocyanine Blue, on the other hand, which evolves as the solvent is removed by distillation, contains so many impurities that it must be boiled before being utilized further.
Phthalic anhydride and urea, together with copper(I) chloride and ammonium molybdate, are heated to 200 °C in trichlorobenzene. The ratios between the components are the same as in the baking process. Carbon dioxide and ammonia are released to yield Copper Phthalocyanine Blue. The reaction is complete after 2–3 h, producing a yield between 85% and >95%.
The main disadvantage of working in the presence of a solvent is the problem of regenerating this solvent.
Apart from nitrobenzene, trichlorobenzene in particular was the preferred solvent up until a few years ago. It has now been replaced by other solvents such as high-boiling hydrocarbons (kerosene, naphthalene) and also alcohols and glycols, because traces of polychlorinated biphenyls may be formed. These are not easily degradable. With hydrocarbons, however, the possibility of fire and explosion must be considered in designing suitable production units.
In the presence of a solvent, the crude pigment generally evolves in much purer form than if it is prepared by baking. Higher degrees of purity, that is, up to 98%, are achieved by additional alkaline and/or acidic treatment. Commercially available types of crude Copper Phthalocyanine Blue typically contain more than 90% pure pigment. This preliminary product is also commonly supplied to and finished by companies who do not manufacture crude pigment themselves.
Reaction between phthalic anhydride and urea always affords chlorine-free Copper Phthalocyanine Blue. Chlorinated derivatives are obtained only in the absence of bases (ammonia) or urea. The phase-stabilized α-modification is prepared by essentially the same but slightly modified route: it is derived from mixed condensation with chlorinated phthalic anhydrides (such as 4-chlorophthalic acid) (Section 3.1.2.4).
Despite the described disadvantages, but due to the high yield, economic production methods, and low-cost starting materials, the phthalic anhydride/urea method is presently the most significant industrial route to Copper Phthalocyanine Blue manufacture.
The gross reaction equation for the preparation of Copper Phthalocyanine Blue from phthalic anhydride and urea may be written as follows:

This overall reaction equation points to the fact that copper phthalocyanine formation formally requires two reduction equivalents.
Formally, copper phthalocyanine is formed by the following reaction pathway:

Phthalic anhydride initially reacts with ammonia, which in turn is liberated, for instance, by decomposition of urea. Diiminophthalimide is then produced via phthalimide and monoiminophthalimide. Subsequent self-condensation (as in the phthalonitrile process) under cleavage of ammonia affords polyisoindolenines, which form complexes with copper ions. Ring closure is achieved through further release of ammonia, and copper phthalocyanine is finally obtained by reduction.
The above-mentioned reaction mechanism is validated by proven intermediates.
Urea acts not only as an ammonia source but also forms decomposition products, such as biuret and higher condensation products. 14C labelling has indicated that the carbon atom of the urea molecule is not incorporated into the phthalocyanine structure. Employing a phthalic anhydride molecule bearing one radioactively labelled carbonyl function affords labelled copper phthalocyanine and phthalimide (as a side product), while the liberated carbon dioxide was found not to show any radioactivity. Labelled carbon dioxide, on the other hand, has been obtained in corresponding experiments using 14C labelled urea.
3.1.2.3 Manufacturing the Different Crystal Modifications
Unsubstituted Copper Phthalocyanine Blue is polymorphous. X-Ray diffraction diagrams point to eleven different crystal modifications (α, β, γ, δ, ɛ-, R, π, ρ-, σ, ζ, and a new eleventh one) [14, 15]. The five important ones are the α-, β-, γ-, δ- and ɛ-phases (Figure 3.1). Their relative thermodynamic stability decreases in the order: β > ɛ > δ > α ≈ γ [16–19]. The crystal structures are described in Section 3.1.3.

Figure 3.1 Copper Phthalocyanine Blue: X-ray powder diffraction diagrams of different crystal modifications, measured in transmission mode with Cu-Kα radiation.
Amongst these modifications, it is particularly the phase-stabilized α-form and the β-form that are of high commercial interest.
Crude Copper Phthalocyanine Blue prepared by the phthalonitrile or urea process typically evolves as the β-modification with a coarse particle size.
The synthesis of the crystal modification is controlled primarily by the finishing technique of the crude pigment. There are basically two different methods to produce a finely dispersed pigment: treatment with acid to form copper phthalocyanine salts, followed by precipitation in water, on the one hand, and mechanical treatment (milling, kneading) on the other hand. The following methods are used:
- dissolving the crude pigment in concentrated sulfuric acid;
- swelling in 50–90% sulfuric acid;
- dry milling in the presence or absence of water soluble salts, as well as with or without small amounts of organic solvents;
- milling in organic solvents;
- kneading with salt in the presence of solvent.
Crude Copper Phthalocyanine Blue has become a commodity product, which is synthesized in large amounts. The finishing process is more demanding. Today, salt kneading and activation by ball milling with subsequent solvent finishing with additives play a major role. An alternative is acid pasting, that is, swelling in sulfuric acid followed by precipitation with water in the presence of amines or additives.
3.1.2.3.1 α-Modification
Dissolving or swelling of crude Copper Phthalocyanine Blue in sulfuric acid, followed by precipitation in water (hydrolysis) affords the α-modification with a fine particle size. Emulsifiers may be present if desired. Dry milling of the crude β-crystal phase, for instance in the presence of sodium chloride or sodium sulfate, also yields the α-phase.
3.1.2.3.2 β-Modification
The β-modification as a rule evolves as a more coarse-grained material than the α-phase. It is prepared by milling the crude Copper Phthalocyanine Blue with salt in the presence of a ‘crystallization stimulating’ solvent. Aromatic hydrocarbons, esters or ketones are normally used.
A primarily platelet-shaped β-modification may also be obtained by the phthalonitrile process if particularly pure phthalonitrile is employed [20].
3.1.2.3.3 γ-, δ-, ɛ-Modifications
Crude Copper Phthalocyanine Blue is stirred in 60% sulfuric acid and the thus obtained sulfate hydrolysed with water. Subsequent filtration affords the γ-phase. It is also possible to knead crude Copper Phthalocyanine Blue with salt (sodium sulfate), concentrated sulfuric acid and a third agent, which may be an alcohol, a polyalcohol, or one of the corresponding organic esters [21]. A third option is to stir α-Copper Phthalocyanine Blue with 30% sulfuric acid and glycol monobutylester or the corresponding ethyl ester or tetrahydrofuran [22].
The δ-form can be obtained by treating Copper Phthalocyanine Blue in benzene or toluene with aqueous sulfuric acid in the presence of a surfactant [23].
The ɛ-phase is produced by comminution of the α-, γ- or δ-modification, for instance in a planetary ball mill. The mill base is then aftertreated in an organic solvent at elevated temperature. It is important to realize that the temperature, depending on the solvent, must be kept below the transition temperature at which the ɛ-phase converts into the β-modification (30–160 °C). The ɛ-modification is made best from the γ-phase, and the most preferred solvents are alcohols [24]. For the industrially hitherto insignificant forms R, π, ρ, σ, and ζ see [1] (Vol. II, 34–35) and [25].
3.1.2.4 Phase- and Flocculation-Stabilized Copper Phthalocyanine Blue Pigments
There are various methods of ‘stabilizing’ a pigment to prevent conversion into a different phase (i.e. change of modification) and flocculation during pigment application. The two following techniques have been found to be most effective:
- minor chemical modification of the Copper Phthalocyanine Blue molecule, for instance by ‘partial chlorination’;
- admixing other agents to the Copper Phthalocyanine molecule; surface active additives may thus help to stabilize the surface.
Only a minor amount of chlorinated copper phthalocyanine, for instance, especially in the 4-position of the copper phthalocyanine molecule, prevents a change of modification from α into β. Approximately 3–4% chlorine is commonly used, which corresponds to the formula CuPc-Cl0.5, also referred to as ‘semi-chloro-CuPc’. The phthalic anhydride/urea synthesis, for instance, affords a partially chlorinated product if 4-chlorophthalic anhydride is added to the reaction mixture. Copper chlorides in the phthalonitrile process have the same effect.
Partial sulfonation or sulfamide formation may stabilize a pigment toward flocculation. Combining partial sulfonation and chlorination will further improve the effect of each individual type of modification. Similar results are observed if carboxylic acid groups are introduced into the pigment molecule.
There is an interesting technique that makes it possible to introduce carboxylic acid groups into a copper phthalocyanine structure by an economic route. Carrying out the phthalic anhydride/urea process in the presence of a small amount of trimellitic acid or another benzene polycarboxylic acid will afford a carboxylated pigment.
Flocculation may likewise be prevented through partial introduction of dialkylaminomethylene groups into the aromatic ring system of the copper phthalocyanine molecule. The copper phthalocyanine structure tightly attaches the basic groups to the surface of the pigment particles:
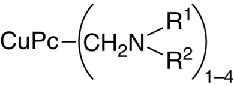
a feature that imparts partial solubility on the parent molecule. Acidic constituents within the binder may interact with the amino groups.
There are other metal complexes, such as tin, aluminium, magnesium, iron, cobalt, titanium and vanadium complexes, which are similarly useful in stabilizing a particular phthalocyanine modification. Moreover, carboxy, carbonamido, sulfo or phosphono-copper phthalocyanine may be admixed during fine dispersion of the pigment.
The discussed additives, which stabilize a pigment with regard to change of modification and flocculation, are usually applied at a concentration of 3–10%.
A flocculation-resistant β-form is obtained by milling crude Copper Phthalocyanine Blue with salt in the presence of xylene or a similar hydrocarbon. In this case, as in general, impurities in the crude Copper Phthalocyanine Blue prevent flocculation.
Polyhalogenated green copper phthalocyanine pigments are not polymorphous and are thus exempt from change of modification.
3.1.2.5 Manufacture of Green Types
The early days of Copper Phthalocyanine Green synthesis were dominated by two competitive routes. One method was the synthesis of tetraphenyl copper phthalocyanine (Bayer), while the second method involved chlorination of copper phthalocyanine in carbon tetrachloride to form copper tetradeca to hexadecachloro phthalocyanine (BASF). It was on the grounds of economic considerations that manufacturers began to prefer the chlorination technique in industrial-scale production.
The oldest and still most prevalent large-scale synthesis for green copper phthalocyanine pigments proceeds via direct chlorination of copper phthalocyanine (1935). Chlorination is effected at 180–200 °C in a sodium chloride/aluminium chloride melt (eutectic blend) in the presence of a catalyst (metal chlorides such as iron(III)chloride). The temperature limits for chlorination are generally reported to be between 60 and 230 °C. There is also a battery of other media, such as chlorosulfonic acid, thionyl chloride, sulfuryl chloride and chlorinated hydrocarbons, possibly under pressure, which are equally suitable for chlorination. All these techniques afford mixtures of multiply chlorinated copper phthalocyanines. The structure is typically chlorinated in at least eight positions, but more often 14–15.5 times. The perchlorinated copper phthalocyanines produced either in AlCl3/NaCl or in acid are then precipitated in water, filtered off, and washed, especially to remove aluminium salts. Since the pigment already evolves from the synthesis as an extremely fine powder, it may then be subjected directly to thermal aftertreatment in an aqueous/organic medium. This is attributed to the fact that perchlorinated copper phthalocyanine interacts with aluminium chloride as well as with strong acids, leading to the formation of salts, which are cleaved through hydrolysis.
Less widely used methods include acid pasting, that is, dissolving the material in a medium such as chlorosulfonic acid and precipitating it in water, or milling the crude pigment. These methods gain importance whenever phthalocyanine is to be chlorinated by a route other than with aluminium chloride or acids.
Commercially available Copper Phthalocyanine Green types typically contain approximately 15 chloro atoms per molecule, while yellowish green types bear not only chloro but also bromo substituents. Such derivatives are obtained through mixed halogenation. Chlorinated green copper phthalocyanines can also be prepared from already perchlorinated starting materials. Tetrachloro phthalonitrile, for instance, may be condensed in nitrobenzene with copper(II) chloride to form copper perchloro phthalocyanine. Costly starting materials, however, make this an industrially unattractive route. Tetrachloro-phthalonitrile may be synthesized from phthalonitrile by gas-phase chlorination, a quite demanding process.
The phthalic anhydride/urea process may also be employed to convert tetrachloro-phthalic anhydride into green copper hexadecachloro phthalocyanine by condensation. In this case, titanium or zirconium dioxides, particularly in the form of hydrated gels, are used instead of the molybdenum salts that are used in the phthalic anhydride process [25]. There is a certain disadvantage to the fact that the products lack brilliance and require additional purification.
The exact hue produced by a green copper phthalocyanine is defined only by the degree of halogenation and by the type of halogen used (chloro or bromo substitution) and not by different crystal phases. Only one crystal modification of copper polyhalophthalocyanine has been reported. A higher degree of chlorination is accompanied by a shift of colour from greenish blue to bluish green. A considerable difference is observed with the introduction of approximately the tenth chloro atom. Bromination affords yellowish shades of green.
Introduced in 1959 by GAF, a copper polybromochloro phthalocyanine was the first commercially used mixed halogenated copper phthalocyanine pigment. It was manufactured, as described above, in molten NaCl/AlCl3 with CuCl2 as a catalyst and bromine besides chlorine to afford a sequence of bromination followed by a chlorination reaction. The thus-prepared pigment contains approximately eleven bromine and three chlorine atoms per molecule. In the most yellowish pigments, the degree of halogenation is 11–12 bromo and 4–5 chloro substituents.
Copper Perbromo Phthalocyanine Green may also be obtained from tetrabromo phthalic anhydride by the phthalic anhydride/urea process in the presence of titanium or zirconium catalysts. This route has not yet been introduced on a commercial scale.
3.1.2.6 Metal-Free Phthalocyanine Blue
There are several pathways to metal-free Phthalocyanine Blue (P.B.16):
- by synthesizing suitable unstable ionic metal phthalocyanine salts, such as alkali or alkaline earth salts, with subsequent demetallization by alcohol or acid;
- by direct synthesis from phthalonitrile or amino-iminoisoindolenine;
- by fusing phthalic anhydride with urea.
The first route to preparing metal-free Phthalocyanine Blue involves treating phthalonitrile with the sodium salt of a higher-boiling alcohol, for instance with sodium amylate. The resulting phthalocyanine disodium salt is demetallized by stirring in cold methanol:

Phthalocyanine is likewise obtained by hydrolysing a corresponding calcium or magnesium salt in an acidic medium.
The route that proceeds via phthalonitrile has stimulated interest in connection with semiconductor technology for photoelectric copying, especially with respect to the pure Phthalocyanine Blue that is prepared by this path. An excellent pure product is obtained by heating phthalonitrile in 2-dimethylaminoethanol while passing ammonia through the reaction mixture. A second scheme involves heating 1,3-diiminoisoindolenine in the same solvent to 135 °C, a method that affords the product not only in equal purity but also in high yield (approx. 90%). A third route proceeds via phthalonitrile, which is heated with hydrogen under pressure in an inert solvent [26]. For dyeing, phthalocyanine as pigment can also be formed on cellulosic fibres itself [27].
The similarly blue metal-free phthalocyanine is chemically somewhat less stable than its copper complex [28]: it decomposes slowly in a sulfuric acid solution. On the other hand, it can be chlorinated to afford metal-free Phthalocyanine Green.
3.1.3 Crystal Structures and Properties
While copper phthalocyanine blue pigments range in shade from greenish to reddish blue, the copper phthalocyanine green series covers the bluish to yellowish green portion of the spectrum. All copper phthalocyanines exhibit excellent fastness properties, especially regarding lightfastness and weatherfastness, and most types show very good heat stability. The pigments do not melt. Copper Phthalocyanine Blue may be sublimed in an inert gas atmosphere at approximately 550 °C and ambient pressure.
The first crystal structures of phthalocyanines were determined by Monteith Robertson as early as in 1935 to 1936 (including β-CuPc, CoPc, NiPc, PtPc and metal-free phthalocyanine) [29, 30, 31]. Actually these crystal structures were amongst the first organic and metal-complex structures ever solved.
Crystal structures of the α- and β-phases of Copper Phthalocyanine Blue polymorphs have been determined by X-ray single-crystal structure analyses (Figure 3.2) [32–34]. The crystal structures of the γ- and ɛ-phases were solved from X-ray powder data and refined using a combined Rietveld and energy-minimization method [15, 35]. In all polymorphic forms, the planar and almost square phthalocyanine molecules are arranged like rolls of coins, that is, in one-dimensional columns (stacks). The molecules are not perpendicular to the stacking direction, but inclined against it. The polymorphs vary in the inclination of the normal vector of the molecular planes against the column direction (α: 25°, β: 46°, γ: 27°, ɛ: 49°), and in the arrangement of the columns. Molecules of neighbouring columns can be parallel (α-phase) or form a herringbone packing (β-, γ- and ɛ-phases) (Figure 3.2).

Figure 3.2 Crystal structures of Copper Phthalocyanine Blue polymorphs. The molecular columns run in the vertical direction.
The distances between the planes of neighbouring molecules within the columns are similar in all polymorphs (α 3.44 Å, β 3.35 Å, γ 3.38 Å, ɛ 3.28 Å). When viewing perpendicular to the molecular planes, it is apparent that neighbouring molecules are situated not exactly on top of each other, but shifted sidewards. The amount and the direction of this shift strongly depends on the polymorphic form (Figure 3.3).
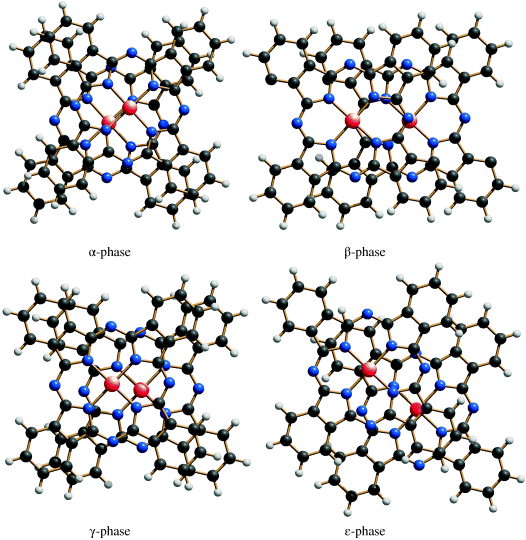
Figure 3.3 View perpendicular to the molecular planes in Copper Phthalocyanine Blue polymorphs. Two neighbouring molecules are shown.
Within the columns the molecules are connected by considerable van der Waals (dispersion) interactions, supported by minor Coulomb interactions between the partial charges of the atoms (e.g. between partially positive Cu and partially negative N atoms). The closest distance between Cu atoms and N atoms of neighbouring molecules varies between 3.28 Å (β-phase) and 3.75 Å (ɛ-phase); these contacts are much longer than the intramolecular CuN bond lengths of 1.9–2.0 Å and should not be regarded as a coordination bond, but as van der Waals and Coulomb interactions only.
The columns form a dense rod packing. This arrangement of the columns can even be seen in HR-TEM images (Figure 3.4a). Between the columns there are weak van der Waals interactions of carbon and hydrogen atoms only (Figure 3.4b).

Figure 3.4 (a) HR-TEM image of CuPc, showing three crystallites. The lattice fringes correspond to the molecular columns. Courtesy of Dr. Tatiana Gorelik, University of Mainz. (b) View along the molecular columns in the α-phase of CuPc.
The prismatic faces of the columns are non-polar. The basal planes of the stacks, on the other hand, which incorporate the copper atoms, the nitrogen atoms and the π-electronic systems, exhibit a relatively polar character. On the whole, the surface of the acicular substituted phthalocyanine pigment is largely non-polar, irrespective of the crystal modification. There is consequently very little chance for specific interaction with the surrounding medium. This also explains the very low hydrophilicity of the compounds discussed thus far, a facet that has some impact on the dispersibility of a pigment, particularly regarding surface wetting and stabilization of a dispersed pigment.
Unsurprisingly, therefore, phthalocyanine pigments show a considerable tendency to flocculate. Types that are stabilized towards flocculation, for instance through introduction of polar groups, exhibit much improved properties in commercially important media. This is true for all modifications. The thus-achieved improvement in pigment performance is a particular asset in various types of gravure and flexographic printing inks and in oven drying paints. This stabilization, however, will often adversely affect the stability of a pigment to organic solvents and its fastness to overcoating.
Copper phthalocyanine pigments also demonstrate good overall stability to organic solvents. Several solvents, however, especially aromatics, may cause a change of modification in unstable types or ‘overcrystallization’ in stable varieties. This phenomenon is largely due to the tendency of the more stable phase to nucleate. The particle size of the resulting crystals decreases as the number of nuclei rises. β-Copper Phthalocyanine Blue is the thermodynamically stable modification.
Only one crystalline modification of polyhalogenated copper phthalocyanine green pigments has been reported. Its crystal structure was investigated by high-resolution transmission electron microscopy and electron diffraction (Figures 3.5 and 3.6) [36]. The molecules are tilted against the stacking direction.

Figure 3.5 Electron micrograph of perchlorinated copper phthalocyanine green [36]. Figure reproduced from Reference [36].

Figure 3.6 Proposed crystal structure of perchlorinated copper phthalocyanine green, determined by electron diffraction [36] with subsequent force-field optimization of the molecular geometry (M.U. Schmidt, unpublished). The crystal symmetry is ambiguous.
The degree and type of halogenation and also the ratio between chloro and bromo atoms in a molecule defines not only the colouristics but also, if only marginally, a series of fastness properties.
Metal-free phthalocyanine, H2Pc, exists in seven polymorphs (α, β, γ, κ, τ, τ′, τ″) [36a]. The crystal structure of the α-phase [36b] is shown in Figure 3.7. It is isostructural to the γ-phase of CuPc. The β-phase of H2Pc [29, 36c, 36d] is isostructural to the β-phase of CuPc. The crystal structure of the phase X of H2Pc, which has gained much interest as a photogenerator, has been investigated by combinations of X-ray powder diffraction, lattice-energy minimisation, electron diffraction and neutron diffraction [36d, 36e]. However, the results remain inconsistent

Figure 3.7 Crystal structures of metal-free phthalocyanine. (a) Molecular structure. In the crystal the two central hydrogen atoms are disordered, i.e. they are attached to the four central N atoms with probabilities of 0.5 each. The diffraction data do not reveal, if the disorder is dynamic (i.e. changing with time), or static (i.e. the individual molecules have different H atom positions). (b) Crystal structure of the α-phase. The β-phase is isostructural to the β-phase of CuPc, see Figure 3.2.
Cobalt phthalocyanine exhibits a crystal structure that is isotypic to the β-phase of copper phthalocyanine.
In the aluminium complex of phthalocyanine, P.B.79, the Al3+ is not only coordinated to the phthalocyanine ligand but also to a chlorine atom (Figure 3.8). Hence, the stoichiometry is ClAlPc.

Figure 3.8 Molecular structure of P.B.79, chloro-phthalocyaninato-aluminium(III) [37].
The crystal structure of P.B.79 (ClAlPc) was determined from single-crystal data [37] with subsequent lattice-energy minimisations by dispersion-corrected density-functional theory [37a]. The molecules are arranged in layers (Figure 3.9). Within the layers the packing of the phthalocyanine moieties is similar to those in the β-phase of CuPc (Figure 3.4b). Molecules of the neighbouring layers have inverted orientations, see Figure 3.10(a). This arrangement is energetically favourable and leads to a very dense packing (Figure 3.10(b)). The resulting double layers are affected by a stacking disorder, see Figure 3.11 [37a].

Figure 3.9 Crystal structure of Pigment Blue 79, ClAlPc. Molecular arrangement in the layer. Al atoms pink, Cl atoms green (behind the Al atoms), N atoms blue, C atoms black, H atoms white.

Figure 3.10 Crystal structure of Pigment Blue 79, ClAlPc, showing one reference molecule with the Cl atom “up”, and three of the surrounding molecules with Cl atoms “down”. (a) Ball and stick model. (b) Space-filling model.

Figure 3.11 Stacking disorder of the double layers in Pigment Blue 79. Starting from the 1st double layer, subsequent double layers are laterally shifted either to the left, or, by a different amount, to the right. This results in an irregular stacking sequence. The drawing shows a typical section of this sequence [37a].
Phthalocyanine pigments, which show high tinctorial strength, provide an excellent ratio of strength versus price. The strongest member is α-Copper Phthalocyanine Blue, while yellowish Copper Phthalocyanine Green is the weakest representative.
3.1.4 Application
By far the largest portion of the worldwide phthalocyanine production is targeted for use as organic pigment. The compound is employed almost exclusively in the form of copper phthalocyanine or as one of its halogenated derivatives. Commercial dyes are produced by introducing solubilizing groups, such as one or more sulfonic acid functions, into the phthalocyanine and especially into the copper phthalocyanine host structure. If desired, the system may undergo further chemical modification. The thus-prepared dyes find extensive use in various areas of textile dyeing (they are utilized as direct dyes for cotton), for spin dyeing and in the paper industry.
The five important crystal modifications of copper phthalocyanine blue pigments differ in terms of colouristics. The β-modification provides the most greenish and cleanest shades of blue, the α-form is distinctly redder than the β-modification, and the ɛ-phase is even redder than the α-form. Both γ- and δ-Copper Phthalocyanine Blue have not yet been commercialized.
Thermal treatment converts the thermodynamically unstable modifications into the stable β-form. The same change of modification may be effected in a high-boiling inert solvent, especially in an aromatic one. This should be considered if unstable modifications of Copper Phthalocyanine Blue are to be applied in plastics or in oven drying paints containing aromatics. The phase transitions have been studied extensively [16]. Minor chemical changes to the copper phthalocyanine structure, for instance through partial chlorination, increase the activation energy for the change of crystal modification, which is a particular concern with the α-phase. If this energy barrier becomes large enough, sufficient stability is imparted on the crystal modification to make it suitable for application in various areas, even in aromatic systems or in polymers. In addition, there are various methods to also improve the stability of these pigments to flocculation (Section 3.1.2.4). Other additives are available that enhance the somewhat poor rheological properties of copper phthalocyanine pigments.
Although low-cost green shades may be produced by combining Copper Phthalocyanine Blue with yellow pigments, the fastness properties of the resulting mixtures are usually inferior to those of green polyhalogenated copper phthalocyanine pigments.
The fully chlorinated copper phthalocyanine is used as precursor for the synthesis of IR-absorbers, applied in optical recording layers for data storage [38].
Metal phthalocyanines with different metals as central atom may become important in several new technologies, such as optoelectronics, optical communication or as catalysts or in medical applications. Part of the new compounds will be represented by pigments [39].
3.1.5 Commercially Available Phthalocyanine Pigments
3.1.5.1 General
The α- and β-types of Copper Phthalocyanine Blue reign supreme amongst commercially available phthalocyanine pigments. There is also increasing interest in the phase-stabilized form of the α-crystal modification. Both modifications are also supplied as flocculation resistant types.
Because phthalocyanine blue pigments are utilized in a host of applications, the requirements for their use vary considerably. Numerous special-purpose types are therefore supplied that are custom designed to exhibit optimized properties in certain media to suit particular demands.
The ɛ-modification has attracted some interest. Commercial types of metal-free phthalocyanine blue are also available.
Copper phthalocyanine green pigments are primarily offered as partially chlorinated products containing 14–15 chlorine atoms per pigment molecule (Pigment Green 7). Various copper polybromochloro phthalocyanine derivatives featuring 4–9 bromine and 8–2 chlorine atoms per molecule (Pigment Green 36) play a role as yellowish green pigments.
Table 3.1 lists the types of phthalocyanine pigments that are currently supplied.
Table 3.1 Commercially available copper phthalocyanine pigments (if not otherwise mentioned). The molecular formula is shown in Scheme 3.1 (Section 3.1).
| C.I. Name | C.I. Constitution Number | Stabilized to change of crystal phase | Crystal phase | Range of shades | Number ofhalogen atoms | Comments |
| P.B.15 | 74160 | No | α | reddish blue | — | |
| P.B.15:1 | 74160 | Yes | α | greener than P.Bl.15 | 0.5–1 Cl | |
| P.B.15:2 | 74160 | Yes | α | reddish blue | 0.5–1 Cl | Non-flocculating |
| P.B.15:3 | 74160 | — | β | greenish blue | 0a | |
| P.B.15:4 | 74160 | — | β | as P.Bl.15:3 | 0a | Non-flocculating |
| P.B.15:6 | 74160 | Yes | ɛ | very reddish blue | 0a | |
| P.B.16 | 74100 | greenish blue | Metal-free phthalocyanine | |||
| P.B.75 | 74160:2 | — | reddish blue | Cobalt phthalocyanine | ||
| P.B.79 | 741300 | — | greenish blue | Aluminium phthalocyanine, ClAlPc | ||
| P.Gr.7 | 74260 | — | bluish green | 14–15 Cl | ||
| P.Gr.36 | 74265 | — | yellowish green | 4–9 Br, 8–2 Cl | ||
| P.Gr.37 | 74255 | — | greenish blue | 8 Cl | ||
| a Depending on the synthetic route, small amounts of chlorine may be present. | ||||||
3.1.5.2 Pigment Blue 15
Under the name of Pigment Blue 15, the Colour Index lists types of α-Copper Phthalocyanine Blue that are not stabilized towards phase-transfer. Compared to other phthalocyanine blue pigments, Pigment Blue 15 types are reddish blue in shade, tinctorially strong and provide high colour yield and economy in use. Their impact on the market, however, is inferior to that of the corresponding stabilized types, by which they are increasingly being replaced. In many media, P.B.15 types are redder and frequently cleaner than stabilized types, an advantage that is often compromised by less tinctorial strength.
Non-stabilized α-Copper Phthalocyanine Blue is utilized in the printing industry, to a certain extent in oil-based binder systems, such as offset printing inks for packaging and metal deco printing. Under standard conditions, P.B.15 is stable to various organic solvents, such as alcohols, esters, ketones, aliphatic hydrocarbons, toluene, and plasticizers, such as dioctyl phthalate (Section 1.6.3). Ethylglycol or the standard DIN 16 524 solvent mixture, however, are slightly coloured. The prints are equally fast to acids and alkali and show perfect fastness to soap, butter and sterilization. P.B.15 is too reddish to be used in process colours, that is, to produce the standard cyan for three- and four-colour printing. A strong tendency to change its crystal modification in the presence of aromatics makes P.B.15 a doubtful candidate for packaging gravure inks. For the same reason, the pigment is only rarely found in paints.
The fact that unstable P.B.15 types tolerate less than 200 °C is a disadvantage in the colouration of plastics. With increasing temperature, the pigment frequently converts into the β-modification, a tendency that is especially pronounced in polymer materials that contain aromatic rings, such as polystyrene, ABS or PET.
As a result of their reddish shade, P.B.15 types are used to a certain extent to colour polyethylene, which is processed below 200 °C, to produce items such as films. Another suitable medium is PVC, which is commonly treated at moderate temperature. There is a certain disadvantage to the fact that the tinctorial strength of such systems is frequently inferior to that of stabilized Copper Phthalocyanine Blue varieties.
Incorporated in plasticized PVC, P.B.15, like other phthalocyanine pigments, is usually entirely fast to migration. Moreover, it provides excellent lightfastness. P.B.15 also finds use in various types of PUR foam materials as well as in rubber. Its redder and frequently cleaner shade compared to corresponding stabilized types makes it an equally useful pigment for other media. This applies especially for water-based systems. Textile printing, paper mass colouration, paper surface treatment and paper pulp are areas of application suitable for the use of P.B.15 in office articles, including coloured pencils, blackboard chalks for schools and watercolours.
Using P.B.15 in aqueous media may be problematic given that the highly non-polar character (Section 3.1.3) of the pigment may make it necessary to use a considerable amount of dispersants such as oxyethylated phenols, alcohols or alkylsulfonates. Adequate dispersion relies as much on sufficient wetting of the pigment particles as it does on vigorous mechanical shear. Thus, adiabatic heat is frequently developed during pigment processing. Unsubstituted types of α-Copper Phthalocyanine Blue may convert into the β-modification if they are treated in insufficiently cooled dispersion units. This process is associated with a colour change to a greener blue as well as with loss of cleanness and tinctorial strength. P.B.15 is thus supplied mainly in the form of highly pigmented aqueous pastes. Pigment manufacturers may either provide such products themselves or leave this task to specialized manufacturers of pigment preparations.
Introducing suitable substituents into the Copper Phthalocyanine Blue structure or covering the crystal surface with appropriate substances may specifically influence the hydrophilicity or the polarity of the pigment (Section 3.1.2.4). The ease of dispersion and wetting may thus be improved or optimized for certain applications, such as for aqueous media.
3.1.5.3 Pigment Blue 15:1
In the Colour Index, the designation P.B.15:1 refers to a phase-stabilized α-Copper Phthalocyanine Blue. This pigment has gained great commercial importance in almost all areas. Stabilization, however, for instance through partial chlorination, usually causes loss of tinctorial strength and cleanness as well as a colour shift towards a greener blue. Despite this disadvantage, however, stabilized P.B.15:1 types reign supreme amongst Copper Phthalocyanine Blue types as colourants for coatings and paints, packaging printing inks and plastics. They show good resistance to organic solvents. Excellent lightfastness and weatherfastness, superior migration fastness, and high heat stability, along with a reasonable price make these pigments attractive products. There is almost no limitation to pigment use in the above-mentioned areas. Moreover, P.B.15:1 types are often employed in combination with other pigments. Blends with Dioxazine Violet, for instance, provide shades of navy blue, while P.B.15:1 in conjunction with titanium dioxide produces good hiding power. In addition, P.B.15:1 blends are also utilized to brighten white paints, printing inks and other media.
The paint industry employs P.B.15:1 for its excellent fastness properties to colour all types of paints, including automotive refinishes and original automotive finishes, in both solid and metallic shades.
The so-called flop effect is a primary concern in connection with metallic finishes. Products may exhibit a different colour and/or brightness, depending on the direction of incident light and the angle of observation. Flop is an important consideration in automotive finishes because different auto parts, although finished with the same product, may appear in a different colour, depending on the angle at which they are viewed. Differences are particularly visible in locations such as between hood and fender. This phenomenon is attributed to a considerable extent of scattering in the coloured pigment, thus sufficiently transparent pigments present something of advantage. More extensive chlorination of α-Copper Phthalocyanine Blue makes it possible to afford products that exhibit very little flop. Suitable special-purpose products are commercially available.
In practical application, most types of P.B.15:1 are completely fast to overcoating in oven drying systems. The pigments are suitable candidates for powder coatings, for instance for acrylate or polyurethane-based systems, in which they do not exhibit plate-out (Section 1.6.4.1).
The use of P.B.15:1 in various types of paints has for many years been hampered by unsatisfactory flow behaviour. Newer methods involving surface treatment of the pigment with suitable agents have made it possible to improve sufficiently the rheology in certain media to afford pigments that satisfy the use requirements.
Many P.B.15:1 type types demonstrate very good stability to change of modification, a feature that largely eliminates application problems. A sample may be tested for its stability in this regard by refluxing the pigment in toluene or xylene, filtering it, dispersing it in its target paint system, and evaluating the resulting product by colouristic comparison with an untreated but equally dispersed reference sample. The tendency of a product to convert into a different modification may similarly be assessed by storing the pigmented paint at elevated temperature, for instance at 50 °C, for two weeks. The thus prepared dispersion is then compared with a reference system to assess possible colour change.
Incorporated in printing inks, phase-stabilized α-Copper Phthalocyanine Blue, like non-stabilized types, is too reddish to be employed as a process colour for three- and four-colour printing. It is used, however, to a considerable extent in all types of printing inks for special and packaging purposes. The prints are stable to common organic solvents and exhibit perfect fastness properties in special application (Section 1.6.2.3). Metal deco prints withstand up to 200 °C for 10 min or 170–180 °C for 30 min. They may safely be sterilized.
Many types of P.B.15:1 are initially very strong in printing inks. To produce 1/1 SD letterpress proof prints at standard conditions, a pigment concentration of 16–19% will suffice, depending on the type. 1/3 SD prints are produced using 7.5–10% pigment. The resulting prints are entirely fast to light: they score as high as step 8 and step 7 on the Blue Scale, respectively.
The list of applications also includes decorative printing inks for melamine based laminated plastic sheets. Notably, however, P.B.15:1 may not be used in decorative printing inks for polyester-based sheets, because the pigment reacts with accelerators and hardening agents. Moreover, most types of P.B.15:1 are not entirely fast to styrene monomer, which is a frequently used solvent for polyesters.
Incorporated in aqueous systems, the main areas of application for stabilized α-Copper Phthalocyanine Blue are in emulsion paints, pastes for textile printing, and aqueous or water soluble gravure and flexographic printing inks for packaging and wallpaper. The textiles printing industry frequently prefers stabilized over non-stabilized α-types, especially if the resulting product must be fast to dry cleaning, that is, to exposure to halogenated hydrocarbons. Even stabilized Copper Phthalocyanine Blue is frequently worked into aqueous application media in the form of an aqueous paste. Such pastes are concentrated pigment formulations that contain predispersed pigment. In contrast to the handling of non-stabilized types (P.B.15), it is not necessary to exercise particular caution in dispersing P.B.15:1 in the presence of dispersion or wetting agents or particular solvents.
Likewise, P.B.15:1 also lends blue shades to plastics, in which it is incorporated either individually or in combination with inorganic or organic pigments. The stabilized type, like its non-stabilized counterpart, is a pigment with high tinctorial strength in polyolefins. 1/3 SD colourations (1% TiO2) are formulated at 0.08–0.1% pigment, depending on the type. Most grades resist exposure to approximately 300 °C and satisfy almost all demands. Pigment dispersibility, however, is somewhat of a problem. Very few commercial products meet the stringent requirements particularly for use in thin films and similar products. Like other types of Copper Phthalocyanine Blue, P.B.15:1 varieties also affect the shrinkage of partially crystalline thermoplastics to an appreciable extent. They are therefore only marginally suited to application in nonrotation-symmetrical injection-moulded articles, such as bottle crates. Many grades of P.B.15:1 are more or less fast to bleeding in plasticized PVC, depending on the type of stabilization. In this medium, the pigment is also very lightfast and weatherfast. Incorporated in rigid or impact resistant PVC, stabilized α-types are less fast to long-term outdoor exposure than β-Copper Phthalocyanine Blue or Green.
P.B.15:1 is also applied in polystyrene, polyamide, polycarbonate (in which it is heat stable up to 340 °C), PUR foam materials and cast resins. Notably, however, the hardening of cast resins that are based on unsaturated polyesters is usually much retarded.
Problems may arise if P.B.15:1 is to be used in natural rubber, because the presence of free copper affects not only the vulcanization process, but also considerably affects the fastness of the product to ageing. P.B.15:1 is thus considered a rubber poison. The free copper content in a pigment should therefore never exceed 0.015%. Commercial types are available that have been tested accordingly.
P.B.15:1, like other types of Copper Phthalocyanine, finds extensive use in the spin dyeing of polypropylene, polyester, polyamide, secondary acetate, viscose rayon and spun rayon. In these, as in other media, P.B.15:1 is very lightfast, and its textile fastness properties are almost entirely if not entirely satisfactory.
3.1.5.4 Pigment Blue 15:2
Several α-Copper Phthalocyanine types that are stabilized towards flocculation and change of modification are registered in the Colour Index as P.B.15:2. Their main area of application is in paints. They are employed wherever P.B.15:1 types show too much of a tendency to flocculate in application or where economic considerations make P.B.15:2 appear more attractive. In accordance with its good fastness to flocculation, P.B.15:2 demonstrates good resistance to rub-out [40] in most common binder systems. Gradual improvement of the flocculation behaviour has been achieved by suitably adjusting the particle size or particle size distribution as well as the shape of the pigment particles. However, stability to flocculation is imparted on these types primarily through chemical modification or through adsorption of suitable agents. The chemically induced resistance to flocculation, however, is frequently associated with a certain loss of fastness to overcoating, especially in systems containing aromatics. For this reason, flocculation resistant types have been introduced which largely eliminate this problem.
Other fastness properties in application largely equal those of P.B.15:1. In printing inks, P.B.15:2 is employed mostly in special gravure and flexographic inks. This is an area in which lack of fastness to overcoating is frequently of no consequence. P.B.15:2, like other α-Copper Phthalocyanine Blue types, is too reddish to be used as a standard cyan for three- or four-colour printing.
3.1.5.5 Pigment Blue 15:3
P.B.15:3, the β-modification of Copper Phthalocyanine Blue, affords a clean shade of turquoise. Pigments of this type are used primarily in graphical printing as well as in finishes and paints, plastics and rubber, textile printing and other areas, such as office articles.
The commercial importance of P.B.15:3 is reflected in the wide variety of available grades, which cover a range of colouristic and application properties. Differences amongst application media are particularly noticeable in aspects such as dispersibility, tinctorial strength, shade and transparency.
The printing inks industry uses P.B.15:3 especially as a blue component on different colour scales for three- and four-colour printing. The pigment corresponds to the CIE 12-66 standard shade of cyan on the European Colour Scale for offset and letterpress prints (Section 1.8.1.1). Also the printed edition of this book is printed with P.B.15:3.
Resinated types are supplied for use in so-called oily binder systems for offset printing inks. However, these are much fewer in number than corresponding P.R.57:1 types, which are used to produce the standard shade of ruby, or hydrazone yellow pigments, which are used for standard yellow.
β-Copper Phthalocyanine Blue is one of the tinctorially strong pigments, although its strength is approximately 15–20% below that of α-Copper Phthalocyanine Blue. At standard conditions, 1/1 SD letterpress proof prints are formulated at 16% of a tinctorially strong type of P.B.15:1, but require approximately 21% β-Copper Phthalocyanine Blue. 1/3 SD prints contain, respectively, 7.5% and 9% pigment. To produce the standard shade of cyan, inks containing 14–15% of a tinctorially strong grade are used.
The prints exhibit excellent application properties. They are, for instance, entirely fast to organic solvents, soap, alkali and acids. They are also fast to sterilization. Metal deco prints demonstrate very good heat stability. The products withstand exposure to 200 °C for 10 min or to 180 °C for 30 min. Although not quite as fast to heat as halogenated types of Copper Phthalocyanine Green, P.B.15:3 is thus somewhat more heat stable than stabilized α-Copper Phthalocyanine Blue.
The fact that P.B.15:3 provides good dispersibility has met with considerable appreciation. This development results from using more economic but less effective dispersion units (such as agitated bead mills) to produce web offset inks today. Excellent solvent fastness renders P.B.15:3 largely insensitive to high processing temperatures, which affect neither the tinctorial strength nor the transparency of the system, the latter being of considerable concern. This is in contrast to the yellow pigments that are employed in three-colour printing and which are accordingly processed at lower temperature. On the other hand, there is some danger that flocculation may occur in systems that are rich in mineral oil, a phenomenon that resembles pigment recrystallization.
In Europe, Copper Phthalocyanine Blue is usually supplied as a powder or as a granulate. The granulated product is somewhat less dusty but also more difficult to disperse. In the USA, P.B.15:3 also continues to be offered in the form of flushed pastes to be incorporated into oil-based printing inks. These pastes offer improved pigment dispersion and frequently afford more glossy and transparent prints.
As in coatings and paints, β-Copper Phthalocyanine Blue tends to flocculate if employed in publication gravure printing inks that contain a high content of aromatics and very little resin. Therefore, nonflocculating P.B.15:4 grades are becoming increasingly important in this area. Sometimes, also used are predispersed, solvent-containing pastes (toluene) or concentrates, nonaqueous dispersions (NADs, Section 1.8.2.2). These systems confer better stability toward flocculation and tinctorial strength on the consequently improved pigment.
P.B.15:3 is usually also offered as a powder, to be used in packaging gravure printing inks. β-Copper Phthalocyanine Blue, like other pigments, is also supplied in the form of nitrocellulose chips or other preparations, for instance on vinyl chloride/vinyl acetate mixed polymer basis, ethyl cellulose or polyvinyl butyral. These pigment preparations are used to advantage where superior transparency, high cleanness of shade and optimized gloss are prime considerations.
Pigment preparations are more commonly used in aqueous/alcohol flexographic printing inks than in other types of inks. Aqueous/alcohol systems present somewhat of a problem, since hydrophobic phthalocyanine pigments are very difficult to adequately wet and disperse. Stabilized α-Copper Phthalocyanine Blue types, which provide a redder shade, dominate throughout the coatings and paints industry. Despite this fact, β-Copper Phthalocyanine Blue types are also useful, although often in flocculation resistant form (see P.B.15:4). They colour industrial paints, architectural paints and emulsion paints. The β-phase, like the stabilized α-crystal modification, is also excellently lightfast and weatherfast in these media.
P.B.15:3, the stable crystal modification of Copper Phthalocyanine Blue, demonstrates excellent heat stability. Most commercial types are therefore entirely suitable candidates for the pigmentation of plastics. β-Copper Phthalocyanine Blue, however, like the α-modification, often presents dispersion problems, especially in polyolefins. As a result of these difficulties, P.B.15:3 is usually employed in the form of pigment preparations that contain predispersed pigment. Moreover, β-Copper Phthalocyanine Blue types, like α-types, tend to nucleate in polyolefins, a problem that may lead to distortion and stress cracking in injection-moulded parts. Good stability to organic solvents, plasticizers and so on, makes β-Copper Phthalocyanine Blue also a useful pigment for plasticized PVC. The resulting pigmented systems are fast to bleeding, although somewhat less so than halogenated Copper Phthalocyanine Green types. The pigment has equally excellent lightfastness and weatherfastness in plastics. Incorporated in rigid PVC, for instance, β-Copper Phthalocyanine Blue is one of the most stable organic pigments known. It is only slightly less fast than halogenated Copper Phthalocyanine Green types. Besides, P.B.15:3 is also found in polystyrene, impact resistant PS types, ABS and similar polymers. Transparent P.B.15:3 systems, like those containing α-Copper Phthalocyanine Blue and Copper Phthalocyanine Green types, are thermally stable up to 300 °C. In very light white reductions (0.01% pigment/0.5% TiO2), however, the samples only withstand up to 250 °C, while corresponding green types (P.Gr.7) are stable up to 300 °C.
β-Copper Phthalocyanine grades are also supplied as special-purpose types with a limited or defined free copper content, targeted for the colouration of rubber (see Section 3.1.5.3).
P.B.15:3, like stabilized α-Copper Phthalocyanine Blue, markedly affects the hardening of unsaturated polyester cast resins. The list of applications also includes PUR foam materials, office articles, such as coloured pencils, wax crayons and watercolours, as well as spin dyeing of polypropylene, polyacrylonitrile, secondary acetate, polyamide, polyester and viscose. Used in polyester spin dyeing, P.B.15:3 satisfies the thermal requirements of the condensation process (Section 1.8.3.8). 1/3 and 1/25 SD samples equal step 7–8 on the Blue Scale for lightfastness. Textile fastnesses, such as stability to wet and dry crocking, are perfect.
3.1.5.6 Pigment Blue 15:4
Under the designation P.B.15:4, the Colour Index lists β-Copper Phthalocyanine Blue types that are stabilized towards flocculation. These products show largely the same colouristic and fastness properties as P.B.15:3 types, but often exhibit much better rheology. As with stabilized α-Copper Phthalocyanine Blue types, stabilization through surface treatment has proven to decrease the solvent fastness of β-Copper Phthalocyanine Blue, sometimes considerably so, making the pigment more sensitive to aromatics, alcohols, ethylene glycol and ketones.
Stabilized types of β-Copper Phthalocyanine Blue are becoming increasingly important throughout the printing inks and paints field. Improved stability to flocculation and other rub-out phenomena [40] make P.B.15:4 an attractive choice, especially for oven drying paints. The pigments have also gained importance in gravure printing inks, particularly in toluene-based publication gravure printing inks and in various types of flexographic printing inks. They now dominate in these areas. Often, P.B.15:4 types are optimized for use in specific media, such as publication gravure printing inks. In these media the pigment exhibits high tinctorial strength and good flow. Other products have been developed for processing in particular dispersion units.
3.1.5.7 Pigment Blue 15:6
P.B.15:6, which is the ɛ-modification of copper phthalocyanine, affords the reddest blue shade provided by any copper phthalocyanine blue pigment. It has become possible to sufficiently stabilize these pigments towards change of modification to largely eliminate undesirable colouristic effects in application. Section 1.6.5 illustrates the different processes that simultaneously occur during pigment dispersion if an insufficiently stabilized grade of this modification is used. Oven drying paints containing aromatics clearly undergo a colour change towards greener shades as the dispersion time increases. This phenomenon is attributed to a conversion into β-Copper Phthalocyanine Blue. Recrystallization effects, which accompany this phase transition, markedly decrease the tinctorial strength of the system. Very interesting colouristically, the ɛ-modification is used in colour filters for liquid crystal displays (LCDs) [28]. Corresponding reddish shades of blue may also be produced by combining α-Copper Phthalocyanine Blue types with small amounts of Dioxazine Violet (P.V.23). The shade of ɛ-Copper Phthalocyanine Blue demonstrates noticeably higher cleanness than the equally reddish blue shades provided by P.B.60.
In terms of fastness properties, P.B.15:6 performs like other modifications. In baking enamels, stabilized types are completely fast to overcoating, but they are not entirely fast to bleeding in plasticized PVC.
3.1.5.8 Pigment Blue 16
To date, metal-free phthalocyanine blue pigment has little commercial importance. Used to an appreciable extent as a greenish blue pigment until the 1950s, it was later replaced largely by β-Copper Phthalocyanine Blue. Of the different crystal modifications, only the α-form continues to play a certain commercial role. The pigment is supplied as a non-stabilized type and also as a type that is stabilized towards change of modification. Regular grades, like the unstabilized α-Copper Phthalocyanine Blue P.B.15, lose tinctorial strength if they are exposed to certain solvents and undergo a colour shift towards a greener blue. In several systems these types may present problems due to lack of dispersibility and flocculation stability.
P.B.16 is used especially to produce metallic finishes. Incorporated in acrylate resin systems for this purpose, the pigment is more weatherfast than types of Copper Phthalocyanine Blue. P.B.16 also lends colour to plastics, although to a more limited extent. It is not as heat stable as stabilized α- or β-Copper Phthalocyanine Blue types. P.B.16 is also used for artists' paints. The polymorph X of P.B.16 can potentially be used as a photoconductor on drums for laser printers and photocopying machines.
3.1.5.9 Pigment Blue 75
The colouristic properties of this cobalt phthalocyanine blue pigment are comparable with those of Indanthrone Blue, P.B.60. It affords rather dull reddish blue shades, which are noticeably redder and duller than α-copper phthalocyanine blue.
3.1.5.10 Pigment Blue 79
This aluminium phthalocyanine pigment is a copper-free alternate to copper phthalocyanine. Its chemical composition is given as ClAlPc. The pigment affords a greenish blue shade. It is used for printing inks.
3.1.5.11 Pigment Green 7
P.Gr.7 type pigments provide a bluish green shade. The fact that P.Gr.7 only exists in one crystal modification eliminates the problems associated with the possibility of phase transition.
P.Gr.7, like copper phthalocyanine blue pigments, demonstrates good overall fastness properties. Its lightfastness and weatherfastness, heat stability and solvent stability are superior even to those of the corresponding blue types. Therefore P.Gr.7 is broad enough in scope to be found in all areas of pigment application. P.Gr.7 grades, together with the yellower chlorinated/brominated green types of P.Gr.36, dominate wherever green shades are required that cannot be produced by combining α- or especially β-Copper Phthalocyanine Blue with suitable organic yellow pigments. Introducing halogen substituents into the copper phthalocyanine molecule reduces the tinctorial strength of the product, a tendency that becomes more noticeable as the halogen content and thus the molecular weight of the pigment increases.
The main area of application for P.Gr.7 is in paints.
P.Gr.7 is lightfast and durable enough to satisfy almost all requirements. It is almost entirely fast to overcoating. Moreover, the pigment performs very well in terms of other properties that are of interest in this type of application. As with other copper phthalocyanine pigments, problems may occasionally occur in attempting to disperse the pigment in its medium or to stabilize the pigment dispersion to flocculation. Improved versions, however, have been introduced into the market. Untreated green pigments frequently show a greater tendency to flocculate than grades of Copper Phthalocyanine Blue that have not been stabilized to flocculation.
P.Gr.7 is used in all types of paints, including high grade original automotive finishes. Paints containing P.Gr.7 are completely suitable for exterior application. Incorporated in powder coatings, the pigment performs better than β-Copper Phthalocyanine Blue. Plate-out is not observed (Section 1.6.4.1).
The printing ink industry utilizes P.Gr.7 particularly for packaging printing inks. In this area, however, green shades are frequently also produced by combining the less expensive β-Copper Phthalocyanine Blue with suitable organic yellow pigments.
Prints made of P.Gr.7 are tinctorially weaker than those containing Copper Phthalocyanine Blue types. At standard conditions, for instance, 1/3 SD letterpress proof prints are made of inks formulated at approximately 17% pigment. Only approx. 8–9% β-Copper Phthalocyanine Blue is needed for the same purpose. Phthalocyanine Green demonstrates excellent overall fastness properties in application. Metal deco prints are thermally stable up to 220 °C for 10 min, they are fast to clear lacquer coating and may safely be sterilized. Incorporated in special gravure printing inks, some types of P.Gr.7 may flocculate, a tendency that is also observed in paints and which results in loss of tinctorial strength, reduced gloss and other problems. In contrast to the different types of Copper Phthalocyanine Blue, P.Gr.7 is suited to use in decorative printing for laminated plastic sheets based on polyester. Melamine resin systems, on the other hand, render the pigment phototropic in daylight, a phenomenon that is in contrast to the behaviour of Copper Phthalocyanine Blue. Chemical compounds that undergo a reversible shade change by exposure to light are referred to as being phototropic. The occurrence of phototropicity in a material is limited to a particular portion of the spectrum and may be reversed by irradiating the sample with a source that emits different radiation, usually in the more long-wave region. Irradiating a decorative P.Gr.7 melamine resin-based print with a xenon arc lamp rapidly destroys the pigment, resulting in a dull shade. The lightfastness of such samples equals step 5 on the Blue Scale.
In plastics, phthalocyanine green pigments are also tinctorially much weaker than corresponding blue pigments. More than 0.2% Pigment Green 7, for instance, is required to produce 1/3 SD HDPE samples (1% TiO2). Less than 0.1% β-Copper Phthalocyanine Blue and approximately 0.08% α-Copper Phthalocyanine Blue are needed for the same purpose. The pigment withstands more than 300 °C. Some types of P.Gr.7 influence the shrinkage of HDPE and other partially crystalline thermoplastics at elevated processing temperature much less than do Copper Phthalocyanine Blue types or brominated derivatives. This is an asset in the pigmentation of larger injection-moulded parts.
P.Gr.7 is also difficult to disperse, especially in polyolefins. Easily dispersible products, however, are available. In plasticized PVC, as in other polymers, the pigment is completely fast to bleeding and exhibits excellent lightfastness and durability. P.Gr.7 is one of the most durable organic pigments in rigid PVC and may even be considered for long-term exposure. It is equally suitable for polystyrene, impact resistant polystyrene, and ABS. Its heat stability in polystyrene, even in white reduction, is up to 300 °C, while blue grades only withstand up to 240 °C. Incorporated in unsaturated polyester-based cast resins, P.Gr.7, in contrast to Copper Phthalocyanine Blue types, does not affect the hardening of the medium.
Grades with a limited free copper content are supplied for the pigmentation of rubber. These pigments do not disturb caoutchouc vulcanization and do not affect the resistance of rubber to ageing.
Utilized in spin dyeing, P.Gr.7 lends colour to all types of commercially important fibres. The products demonstrate excellent lightfastness and weatherfastness. Used in polyacrylonitrile, for instance, P.Gr.7 satisfies the stringent requirements for use in outdoor textiles such as canvasses. Its textile fastness properties are almost, if not completely, satisfactory. This textiles field is another area in which Copper Phthalocyanine Blue types are more than twice as strong as P.Gr.7.
Moreover, P.Gr.7 is also found in various applications outside the previously discussed areas, for instance in surface covering colourations for leather and in furniture stains.
3.1.5.12 Pigment Green 36
Pigments of this type provide very yellowish shades of green. They are much yellower than P.Gr.7 types. The colour of P.Gr.36 becomes yellower as more chlorine atoms are replaced by bromine. In the USA, these products are therefore classified according to the yellowness of shade and the bromine content. The bromine content may range from 25–30 wt% for less yellowish to 50–53 wt% for considerably yellower varieties. Again, only one crystal modification has been found.
All types of P.Gr.36 provide excellent lightfastness and weatherfastness, heat stability and solvent fastness. Their main area of application is in paints. The pigment covers the entire range of applications, including various types of high grade automotive finishes. P.Gr.36 types are weaker and more expensive than P.Gr.7 or Copper Phthalocyanine Blue. Yellowish shades of green are thus also produced by combining more inexpensive chlorinated copper phthalocyanine green pigments with suitable organic yellow pigments, although some of the excellent fastness properties are lost in the process.
The list of applications also includes printing inks, in which P.Gr.36 is tinctorially weaker than its copper phthalocyanine counterparts. For comparison, 1/3 SD letterpress proof prints containing P.Gr.36, printed at standardized film thickness, require approximately 26% pigment, while only 17% of P.Gr.7 suffice for the same purpose. The fastness properties exhibited by P.Gr.36 in prints, as in paints, closely resemble those of P.Gr.7, including the phototropic behaviour of decorative printing inks for melamine-based laminated plastics sheets.
More than 0.3% pigment is required to formulate 1/3 SD HDPE samples. P.Gr.36 is thus much weaker than P.Gr.7 types, although it withstands more than 300 °C and is thus equally heat stable. Considerable influence on the shrinkage of injection-moulded polyethylene parts necessitates some caution in using P.Gr.36 for this purpose. Many types are difficult to disperse in plastics, especially in polyolefins.
3.2 Quinacridone Pigments
Strictly speaking, quinacridone pigments are dioxotetrahydroquinolinoacridine compounds. The typical quinacridone structure is a five-ring polycyclic system. The molecule consists of a central benzene ring that is bridged to two peripheral six-membered aromatic rings by two 4-pyridone rings [41]. Quinacridone molecules have been prepared in an angular form, such as in (31, 32), as well as in a linear arrangement, as in (32, 33).

While compounds 31 through 33 are only slightly yellowish, 34 provides an intensely bluish red shade.
Amongst the four constitutions, only 34 has stimulated commercial interest. Systematically referred to as a 5,12-dihydro-quino[2,3-b]acridine-7,14-dione, it is commonly referred to as linear trans-quinacridone. The pigment has gained commercial recognition especially for its high tinctorial strength. Unless mentioned otherwise, the term ‘quinacridone’ is therefore used in this context exclusively to refer to the parent ring system 34.
The first literary evidence of an angular quinacridone (structure 32) dates to 1896, but it was not until 1935 that the quinacridone 34 was first synthesized, by H. Liebermann [42].
Some 20 years later, W.S. Struve (1955) at Du Pont was the first to recognize the impact of linear quinacridone on the pigment industry. In due course, the first industrially useful synthetic pathway was found. Three commercial types of unsubstituted quinacridone consisting of two crystal modifications were introduced in 1958 [43]. From then on, quinacridone pigments experienced rapid development.
3.2.1 Starting Materials, Manufacture and Crystal Structures
Several synthetic pathways for the commercial manufacture of quinacridone pigments have been published. In this context, only those routes are mentioned that were developed for industrial scale production. There are four options, the first two of which are preferred by the pigment industry. Surprisingly, these are the methods that involve total synthesis of the central aromatic ring. On the other hand, routes that start from ready-made aromatic systems and thus might be expected to be more important actually enjoy only limited recognition.
The synthesis found by Liebermann involves heating 2,5-diaminoterephthalic acid with boric acid to 200–250 °C. Ring closure through condensation affords linear quinacridone [42]. A variation of this route, known as acidic ring closure (Section 3.2.1.2), currently plays a major role in pigment manufacture.
3.2.1.1 Thermal Ring Closure
The oldest known industrially used synthesis is carried out in a solvent and involves several intermediate steps. It is not necessary to isolate the intermediates, and the reaction can proceed in one vessel [44].
The starting material is succinic dialkyl ester*, which is easily obtained from maleic anhydride. Cyclization is accomplished with sodium alcoholate in a high boiling solvent or solvent mixture (such as diphenyl ether/diphenyl) to afford the succinylosuccinic dialkyl ester 35:

A newer synthetic pathway, found by Lonza, involves chlorinating diketene to form the γ-chloroacetoacetic ethyl ester, followed by condensation to afford the succinylosuccinic diethyl ester [45].
The typical quinacridone synthesis may be exemplified by the manufacture of unsubstituted quinacridone.
Succinylosuccinic dialkyl ester is treated with two equivalents of aniline to yield the 2,5-dianilino-3,6-dihydroterephthalic dialkyl ester 36. Without separating the intermediate, the ring is closed thermally (at 250 °C) in the same reaction medium to afford the α-modification of dihydroquinacridone 37 [44, 46]:

According to the patent literature, this synthesis proceeds in one vessel. The yield is approximately 75% relative to the starting material succinic dialkyl ester.
The condensation reaction with aniline may also be accomplished in two separate steps. It is possible to separate succinylosuccinic ester, followed by condensation in boiling ethanol in the presence of an acid (acetic acid, hydrochloric acid, phosphoric acid).
Oxidation of dihydroquinacridone to quinacridone may be achieved, for instance, with the sodium salt of m-nitrobenzene sulfonic acid in aqueous ethanol in the presence of sodium hydroxide solution [47]. A distinction is made between heterogeneous and homogeneous oxidation. The reaction is referred to as a ‘solid state oxidation’ if the solvent contains approximately 2% sodium hydroxide solution. A content of approximately 30% sodium hydroxide solution relative to the solvent mixture, on the other hand, converts the reaction into a so-called ‘solution oxidation’. The type of ring closure defines the crystal modification of the resulting dihydroquinacridone, while the oxidation technique defines the crystal phase of the quinacridone pigment.
Solid state oxidation, both of the α- and the β-phase [48] of dihydroquinacridone, affords crude α-quinacridone. Subsequent milling with salt in the presence of dimethylformamide produces the γ-modification, while the β-form evolves in the presence of xylene. Solution oxidation of dihydroquinacridone, possibly performed as air oxidation in the presence of 2-chloroanthraquinone [49], forms crude β-quinacridone. Milling with xylene likewise affords β-quinacridone pigment.
Other patents describing variations of this method have been issued to Du Pont. However, no new insight has yet been gained from these publications of the described processes.
3.2.1.2 Acidic Ring Closure
Acidic ring closure resembles the Du Pont process in that the central benzene ring of the quinacridone structure is synthesized totally. The process bears resemblance to the Liebermann method [42] (see also References [41, 43]). Condensation of succinylosuccinic ester with two equivalents of arylamine affords 2,5-diarylamino-3,6-dihydroterephthalic diester. Subsequent oxidation with suitable agents yields 2,5-diarylaminoterephthalic diester (38). Hydrolysis and cyclization in polyphosphoric acid or other acidic condensation agents produces crude quinacridone [50]. This product already consists of very small particles.
Performing the last step (thermal aftertreatment) in an appropriate solvent provides a simple and convenient method of producing the particular target crystal modification:

The β-crystal modification is prepared by pretreating the crude quinacridone product with alkali base before finishing with the solvent. Immediate solvent treatment, on the other hand, produces the γ-modification of the quinacridone pigment.
The β- and γ-modifications can be directly obtained from the synthesis, if the hydrolysis of the polyphosphoric acid mixture is performed at 135–165 °C with a controlled amount of water or orthophosphoric acid [51, 52].
3.2.1.3 Dihalo-terephthalic Acid Process
Amongst syntheses that start from preformed aromatic systems, the Sandoz process, described here, in particular has stimulated interest. It is the only route that allows the manufacture of asymmetrically substituted quinacridones without obtaining a statistical mixture of symmetrical and asymmetrical molecules. 2,5-dibromo-1,4-xylene or its 2,5-dichloro derivative is obtained by bromination or, correspondingly, chlorination of 1,4-xylene. It is oxidized to form 2,5-dibromoterephthalic acid or its dichloro derivative 40. Subsequent reaction with arylamine, for instance in the presence of copper acetate, affords 2,5-diarylaminoterephthalic acid (41). It is also possible to replace the halogen atoms stepwise by arylamino moieties [53]. Cyclization to form linear trans-quinacridones, as in the above-mentioned method, is achieved by using acidic condensation agents:

Although the synthesis is completed in very few steps, oxidation of 1,4-xylene to the corresponding terephthalic acid does not afford a uniform product. Partial dihalogenation gives rise to side products. The condensation reaction requires two equivalents of arylamine per halogen atom. One equivalent is needed to neutralize the generated hydrohalogen acid, which is subsequently separated as arylamine-hydrohalide and recycled as hydrohalide and arylamine.
3.2.1.4 Hydroquinone Process
The hydroquinone process was developed by BASF [54]. Hydroquinone-2,5-dicarboxylic acid is prepared by a modified Kolbe–Schmidt synthesis from hydroquinone and carbon dioxide. Subsequent reaction with arylamine in an aqueous-methanolic suspension in the presence of an aqueous sodium chlorate solution and a vanadium salt affords the product in good yield:

Subsequent ring closure of the 2,5-diarylamino-1,4-benzoquinone-3,6-dicarboxylic acid (42) is performed in concentrated sulfuric acid (or with thionyl chloride/nitrobenzene) to afford the linear trans-quinacridone quinone 43:

Compound 43 is also a component in a solid solution (mixed-crystal) with quinacridones, types of which are commercially available (Section 3.2.4). Quinacridone quinone is reduced with zinc or aluminium powder in dilute sodium hydroxide solution, in an aluminium chloride/urea melt or in sulfuric/phosphoric acid under pressure to form quinacridone. There are certain disadvantages to this method in view of the fact that hydroquinone is relatively expensive, quinacridone is not easily reduced, and ecological problems prevail (wastewater pollution).
To summarize this discussion, the two latter methods have failed to gain the commercial recognition enjoyed by the first two processes (Sections 3.2.1.1 and 3.2.1.2).
3.2.1.5 Substituted Quinacridones
Certain disubstituted quinacridones are also used commercially. The two peripheral rings in such quinacridone systems (34) are partially chlorinated or methylated. In the sequence 38 → 39 → 34 (Section 3.2.1.2), possible substitution sites on the peripheral rings are indicated by dashes.
Succinylosuccinic diester is cyclized according to the common route by using chlorinated or methylated aniline. Starting from anilines, which are substituted in 2- or 4-position, the reaction affords the symmetrical 4,11- or 2,9-disubstitution products. Reaction with 3-substituted anilines, on the other hand, produces a mixture of both of the symmetrical 1,8- and 3,10-disubstitution products, as well as unsymmetrical 1,10-disubstituted compound. The following chart illustrates this point:

3.2.1.6 Quinacridone Quinone
Quinacridones are not the only industrially significant products. The list may be extended to include the derivative mentioned in Section 3.2.1.4, the linear transquinacridone quinone 43. There are two other synthetic pathways besides the hydroquinone method. The older method involves cyclization of the 2,5-bis-(2-carboxyanilino-)-1,4-benzoquinone (44) with concentrated sulfuric acid or polyphosphoric acid at 150–200 °C. The starting material 44 is obtained through condensation of 1,4-benzoquinone with anthranilic acid:

A more recent synthesis proceeds via 2,5-diarylamino-3,6-dicarbethoxy-1,4-hydroquinone (46), which is obtained by reducing 2,5-diarylamino-3,6-dicarbethoxy-1,4-benzoquinone (45) with sodium hydrogen sulfite or sodium in ethanol:

The hydroquinone 46 is cyclized in an organic solvent at 230–270 °C to yield 6,13-dihydroxy-quinacridone 47. Oxidation with suitable agents (such as nitrobenzene, chromic acid, nitric acid) provides the linear trans-quinacridone quinone (43):

3.2.1.7 Polymorphism
Most quinacridone pigments are polymorphic, that is, they exhibit multiple crystal modifications with different colours and different fastness and application properties. The individual polymorphs are identified by X-ray powder diffraction. The formation of the different polymorphs depends mainly on the procedures used for synthesis and finishing. This effect may be illustrated on unsubstituted quinacridone.
Most synthetic methods afford crude α-quinacridone [55]. This modification, however, has not been commercialized because of its lack of fastness properties. α-Quinacridone may be converted into the β- or γ-phase by organic solvents, especially at elevated temperature. Moreover, the α-phase is much less lightfast and weatherfast than its β- and γ-counterparts. It is by employing suitable solvents, therefore, that the β- and γ-phases are commonly obtained. The list of options includes milling the crude pigment with salt in the presence of a solvent, by first dissolving and then precipitating crude quinacridone, or by heating the crude product. Grinding with salt in a ball mill in the presence of xylene or o-dichlorobenzene, for instance, yields the β-modification [56]. The more stable γ-form [57] evolves in the presence of DMF [58].
The β-form is obtained by dissolving crude quinacridone in any one of various solvents (such as concentrated sulfuric acid/toluene or methylated sulfuric acid), followed by precipitation with water. The same end is achieved by dissolving the product in polyphosphoric acid, followed by rapid precipitation with ethanol at 45 °C. Depending on the process, the β-product, however, may contain some α-crystal modification as well.
The γ-modification is produced by heating crude quinacridone in ethanol (under pressure) or in dimethylformamide or dimethyl sulfoxide at 150 °C.
Non-polar solvents generally aid the formation of the β-phase, while polar solvents assist in producing the γ-modification.
In the patent literature many further phases of quinacridone have been described, including the phases BI [59], γ′ [60], γI [61], γII [62], γIII [63], γIV [64], δ [65], Δ [66], ɛ [67], another ɛ-phase [66] and at least one ζ-phase. All phases were characterized by X-ray powder diffraction. However, X-ray powder diagrams depend not only on the polymorphic form, but also on the crystal sizes, on the crystal quality and on impurities (which may cause additional lines, or distort the lattice and result in modified reflection positions and intensities). Additionally, the diffractometer plays a role. (Most older X-ray powder diagrams have been measured in a reflection mode, which frequently causes incorrect intensities due to preferred orientation effects.) Closer inspection [68] reveals that BI and β actually describe the same phase (β-phase). Similarly, all γ-phases correspond crystallographically to the same polymorph (γ-phase). The δ-phases correspond either to the γ-phase or they consist of a mixture of γ with a trace of β-phase. Additionally the Δ-phase is the same polymorph as γ. The two ɛ-phases, which are claimed to be clearly different from each other, are actually both the γ-phase. The ζ-phase appears to be a mixture of at least three different phases (α, β and γ). One could come to the conclusion that quinacridone exhibits only the three polymorphic forms α, β, and γ, which were already described by Struve in 1958 [55, 56, 58]. However, quinacridone in fact exists in four polymorphs. From the X-ray powder data it is evident that the crude quinacridone, which is usually described as the ‘α-phase’, does not represent a uniform polymorph, but two different polymorphs (or a mixture of them), depending on the synthesis. These two polymorphs, called αI and αII, differ in their X-ray powder diagrams and in their colours (Figure 3.12). The αI-phase corresponds to the phase described by Struve [55] and Labana & Labana [41]. This phase can be obtained by synthesis [55], by milling with NaCl or by sublimation at 140–170 °C [69]. The αII-phase emerges from synthesis under different conditions, or from recrystallization in concentrated sulfuric acid followed by treatment in 3-nitrotoluene at 70–80 °C for 4 weeks [70]. According to Jaffe, the ‘α-phase’ (apparently meaning the αI-phase) offers similar colour attributes as the γ-phase [71], whereas Schmidt describes the αI-phase as a dull reddish-violet powder [68]. The αII-phase is red (Figure 3.12).

Figure 3.12 Colours (a) and X-ray powder diagrams (b) of the four phases of unsubstituted quinacridone. The powder diagrams were measured in transmission geometry, with Cu-Kα1 radiation. (Reproduced from Reference [68] with the permission from The Royal Society of Chemistry.)
The αI- and αII-phases are less stable than the β- and γ-phases, which impedes their use as pigments. Both α-phases are stable as powders at room temperature at day-light for at least 20 years.
3.2.1.8 Crystal Structures
The crystal structure of the β-phase of unsubstituted quinacridone was determined several times by different authors using single-crystal X-ray diffraction [71–74]. The same is valid for the γ-phase [68, 72, 73, 75–79].
For the αI and αII-phases, no single crystals could be grown. The crystal structure of αI-quinacridone was determined from unindexed X-ray powder data by Leusen and Paulus in 1994 [68, 80, 81]. In the first step Leusen predicted the possible crystal structures of quinacridone by global lattice-energy minimizations in various space groups with free lattice parameters. Besides the structures of the β- and γ-phases they found a structure that had a simulated powder diagram similar to the experimental powder diagram of the αI-phase. Subsequent Rietveld refinement by Paulus resulted in the crystal structure of the αI-phase. This was probably the first instance in which an organic crystal structure has ever been solved by a full crystal structure prediction. The structure was independently confirmed by Ogawa using electron diffraction (Figure 3.13) [68, 69].

Figure 3.13 High-resolution transmission electron micrograph of the αI-phase of unsubstituted quinacridone (P.V.19). Photograph kindly provided by T. Ogawa from Kyoto University.
The crystral structure of αII-quinacridone was determined in 2015 by T. Gorelik by means of electron diffraction, using crystals with a thickness of 10 nm only [70]. The diffraction pattern showed strong diffuse streaks through the reflections. Nevertheless Gorelik could solve and refine the structure from the electron diffraction data. The structure was confirmed by lattice-energy optimization with dispersion-corrected density-functional theory calculations (DFT-D).
In all structures of unsubstituted quinacridone (and most structures of substituted quinacridones) the molecules form four CO![]() HN hydrogen bonds (two as donor, two as acceptor). In the phases αI, αII and β the molecules are connected to two neighbouring molecules by two hydrogen bonds each, leading to chains (Figures 3.14a and 3.15a). These chains are stacked on top of each other leading to two-dimensional sheets. The chains themselves are not fully planar, but exhibit small steps between the molecules (Figure 3.15b). In the β-phase the steps have a height of 0.35 Å. Lattice-energy minimizations show that the formation of these steps allows a slightly denser packing of molecules than for exactly planar chains. In the αI-phase all chains are parallel (Figure 3.14b). The structure of the αII is disordered and exhibits a statistical sequence of herringbone arrangements and parallel arrangements of neighbouring chains, with the herringbone arrangement being more frequent (Figure 3.14c). This disorder causes the observed diffuse scattering. The disorder is also the reason, why the structure of the αII-phase could not be found in the crystal structure prediction in 1994.
HN hydrogen bonds (two as donor, two as acceptor). In the phases αI, αII and β the molecules are connected to two neighbouring molecules by two hydrogen bonds each, leading to chains (Figures 3.14a and 3.15a). These chains are stacked on top of each other leading to two-dimensional sheets. The chains themselves are not fully planar, but exhibit small steps between the molecules (Figure 3.15b). In the β-phase the steps have a height of 0.35 Å. Lattice-energy minimizations show that the formation of these steps allows a slightly denser packing of molecules than for exactly planar chains. In the αI-phase all chains are parallel (Figure 3.14b). The structure of the αII is disordered and exhibits a statistical sequence of herringbone arrangements and parallel arrangements of neighbouring chains, with the herringbone arrangement being more frequent (Figure 3.14c). This disorder causes the observed diffuse scattering. The disorder is also the reason, why the structure of the αII-phase could not be found in the crystal structure prediction in 1994.


Figure 3.14 Crystal structure of the αI- and αII-phases of unsubstituted quinacridone (P.V.19). (a) Molecular chains in the αI-phase. The αII-phase is built from the same chains. (b) Packing of the chains in the αI-phase, view along the chains. (c) Packing of the chains in the αII-phase, view along the chains. The crystal structure of the αII-phase is disordered and contains a statistical sequence of herringbone arrangements (H) and parallel arrangements (P) of neighbouring chains.

Figure 3.15 Crystal structure of the β-phase of unsubstituted quinacridone (P.V.19). (a) One chain, surrounded by six chains from neighbouring sheets. (b) Side-view of the chains within one sheet, showing the small steps between the molecules in the chains. (c) Arrangement of chains in neighbouring sheets.
In the β-phase of quinacridone, chains of neighbouring sheets form an angle of 69° (Figure 3.15a,c). This feature is unique for the β-phase; it has not been observed for any other quinacridone pigment, and is rare for other chain structures as well. In the chain direction the molecules interact through hydrogen bonds, in the stacking direction through van der Waals and Coulomb forces, and in the third direction through weak van der Waals interactions between carbon and hydrogen atoms only.
In the γ-phase of unsubstituted quinacridone each molecule is connected to four neighbouring molecules (Figure 3.16a), which leads to a hunter's fence (criss-cross) pattern (Figure 3.16b).

Figure 3.16 Crystal structure of the γ-phase of unsubstituted quinacridone (P.V.19). (a) One molecule and its four neighbouring molecules connected by hydrogen bonds. (b) View along the hydrogen bonds, showing the hunter's fence arrangement of molecules.
The strong intermolecular hydrogen bonds between the CO and the NH-groups combined with the van der Waals and Coulomb forces lead to very stable crystal structures with high lattice energies. This all together causes the very good thermal stability and solvent resistance of these pigments. The solutions, in contrast to the pigment itself, are only poorly lightfast.
In diluted solutions, for example in DMSO (dimethyl sulfoxide) at 180 °C, quinacridone shows a pale yellow to pale orange colour. Quantum-mechanical calculations on an isolated molecule point to a yellow or orange shade as well. The red or violet colour of quinacridone pigments is a solid state effect. The shift from the yellow–orange colour of an isolated molecule to the red or violet shades of quinacridone in the solid state and the colour differences between the individual polymorphs are apparently caused by two effects:
- The formation of the hydrogen bonds causes a weakening of the CO and the NH bonds and an increase of the conjugation between the aromatic rings. In the isolated quinacridone molecule the π-systems of the three benzene rings are only weakly conjugated by the NH and CO bridges; the resulting yellow colour resembles the colour of other substituted benzene compounds (e.g. nitrobenzene). In the solid state the improved conjugation leads to a strong bathochromic shift (Figure 3.17). The different hydrogen bond patterns probably result in different values of the bathochromic shift.
- The colour is affected by exciton interactions (see Section 1.5.3.1) between an excited molecule and all surrounding molecules, including those not connected by hydrogen bonds. The two molecules above and below the excited molecule play a major role. Depending on the polymorphic form, the position of these molecules varies (Figure 3.18). Correspondingly the colours vary.
From Figures 3.14 and 3.15 it is evident that in the chain structures there is no space for any substitution in the positions 1, 4, 8 or 11, that is, in ortho-positions to the NH and CO groups. Any substituent (except hydrogen) in one of these positions would cause negative steric interaction with the neighbouring molecule and impede the formation of a chain structure. The hunter's fence structure of γ-quinacridone tolerates substituents (e.g. Cl) in the positions 1, 4, 8 or 11, if the substituent is not too large. With increasing size of the substituent (H < F < Cl < CH3) the hydrogen bonds are distorted and weakened; correspondingly the lattice energy is reduced and the compounds become increasingly soluble and the lightfastness drops. This explains the orange–red colour and the reduced lightfastness of 4,11-dichloroquinacridone. Even larger substituents in the 4- and 11-positions, for example, tertiary butyl groups, impede the formation of hydrogen bonds even in a hunter's fence arrangement, and the compounds become soluble.

Figure 3.17 Increasing conjugation in solid quinacridone due to hydrogen bonding in the solid state.

Figure 3.18 Stacking of quinacridone molecules. Two molecules, viewed perpendicular to the molecular planes. (a) Unsubstituted quinacridone, β-phase; (b) unsubstituted quinacridone, γ-phase; (c) 2,9-dimethylquinacridone, commercial phase; and (d) 2,9-dichloroquinacridone, commercial phase.
The same effect is observed with substituents on the NH group; for example, N,N′-dimethylquinacridone is the least lightfast and dissolves even in ethanol.
Molecules with substituents in the positions 2, 3, 9 and 10 could, in principle, either form chain structures such as αI-quinacridone or β-quinacridone, or form a hunter's fence structure such as γ-quinacridone. Both cases are observed for 2,9-dimethylquinacridone: The commercial phase exhibits a chain structure such as αI-quinacridone (Figure 3.19) [72, 82, 83], but in addition there exists a second phase of 2,9-dimethylquinacridone, which is described as isostructural to γ-quinacridone [84].

Figure 3.19 Crystal structure of the commercial phase of 2,9-dimethylquinacridone (P.R.122).
The thermodynamically stable phase of 2,9-dichloroquinacridone exhibits a chain structure (Figure 3.20) [72, 85]. The metastable black δ-phase, which is obtained only from sublimation, exhibits a strange structure without any hydrogen bonds [86].

Figure 3.20 Crystal structure of the stable phase of 2,9-dichloroquinacridone (P.R.202).
Pure 3,10-dichloroquinacridone also crystallizes in a chain structure (Figure 3.21) [87]. The structure of the commercial product P.R.209, which contains a mixture of the 3,10-, 1,8- and 1,10-isomers, has not been determined yet.

Figure 3.21 Crystal structure of pure 3,10-dichloroquinacridone (isomer within P.R.209).
Crystal structure data of quinacridone quinone pigments have not yet been published.
3.2.1.9 Solid Solutions
Quinacridones tend to form solid solutions with other quinacridone compounds or with quinacridone quinone. In a solid solution, also called ‘mixed crystal phase’, different molecules adopt a common crystal lattice; on each position within the lattice either a molecule of the one kind or of the other kind is situated, generally with a statistical distribution. Ternary solid solutions exist as well, for example, formed by 2,9-dimethyl-, 2-methyl- and unsubstituted quinacridones. The formation of solid solutions allows the stabilization of less stable polymorphic forms, or the tuning of the molecular packing and thereby of the properties (Section 3.2.4). Solid solutions are identified by their X-ray powder pattern, which is different from the powder pattern of a physical mixture of the individual compounds. Solid solutions can, like the pure pigments, be polymorphic.
3.2.2 Properties
All industrially significant quinacridone pigments are deeply coloured products. They cover the spectral range from yellowish red to violet shades. Quinacridone pigments do not melt but decompose at high temperatures. In vacuum quinacridones can be sublimed, which can be used for example in the production of optoelectronic devices.
Quinacridone pigments are practically insoluble in most common solvents and provide extensive migration resistance in all application media. They are very lightfast and weatherfast.
Colour and properties may be influenced by:
- changing the polymorphic form,
- changing the particle size,
- introducing substituents into the ring system,
- forming solid solutions (mixed crystal phases), for example, of quinacridone with substituted quinacridones or with quinacridone quinone.
The effect of the polymorphic form, that is, of the crystal structure, is the strongest one. Crystal structures, substitution patterns and their implication on the pigments' properties have been described in Section 3.2.1.8. Solid solutions are described in Section 3.2.1.9.
3.2.3 Application
Good-to-excellent fastness properties justify the comparatively high price of quinacridone pigments. They are thus suitable candidates for the pigmentation of high grade industrial finishes. Systems containing quinacridone pigments include original automotive finishes and automotive refinishes, weatherfast emulsion paints such as house paints, plastics, high grade printing inks for purposes such as metal deco and poster printing, and weatherfast textile printings, as well as spin dyeing.
3.2.4 Commercially Available Quinacridone Pigments
3.2.4.1 General
Although quinacridone pigments were first prepared in 1935, their technical value initially went unrecognized. It was only when their polymorphism was detected that quinacridones gained importance. The systematic synthesis of different crystal modifications by appropriate aftertreatment of crude quinacridone made it possible to obtain various highly stable pigments, which rapidly gained commercial interest as a new class of pigments.
A considerable number of mono- to tetra-substituted quinacridones has been described so far. A publication that only includes the literature up to the year 1965 already lists more than 120 compounds, and many exhibit more than one crystal modification [88]. However, this diversity of compounds does not reflect commercial variety. Very few species have been offered, and even less command a major share of the pigments market.
Heading the list are two crystal modifications of Pigment Violet 19 (the parent compound of the quinacridone series). Both the red-violet β- and the red γ-modification are encountered in the market, while the pure α-forms are not lightfast and durable enough to have any commercial value. However, traces of the α-modifications are found in various types.
By addition of 0.1–10% of 2,9-dichloroquinacridone to β-quinacridone as well as to γ-quinacridone during the finishing process, 2,9-dichloroquinacridone acts as an inhibitor for the α-phase. Without α-quinacridone the colour of the β-quinacridone shifts to a bluer violet shade and the colour of the γ-quinacridone to a bluer red shade.
Optionally, 4,11-dichloroquinacridone can be added together with 2,9-dichloroquinacridone to the finishing processes of β- or γ-quinacridone. The incorporation of 4,11-dichloroquinacridone with the 2,9-dichloroquinacridone not only inhibits the formation of the α-phase, but the formed milled products show also increased heat stability properties [89].
Of the substituted quinacridone systems, it is primarily the 2,9-dimethyl derivative (Pigment Red 122) which enjoys appreciation for its excellent fastness properties and its pure bluish red (magenta) shade. In addition, the list also includes 2,9-dichloro-, 3,10-dichloro-, 4,11-dichloro- and 4,11-dimethyl-quinacridones. Some of these are available as solid solutions with other quinacridone derivatives [91].
Other solid solutions are made from quinacridone quinone with unsubstituted quinacridone or 4,11-dichloroquinacridone as a second component.
Quinacridone quinone itself (Section 3.2.1.6) is a tinctorially relatively weak yellow compound with poor lightfastness. Formation of solid solutions with other quinacridones (Section 3.2.1.9) as well as treatment with various metal salts [92] leads to an improvement of light- and weatherfastness. Oxidation of dihydroquinacridone with less than molar amounts of chromate affords a quinacridone/quinacridone quinone solid solution that offers an interesting shade of gold.
Table 3.2 lists representative commercial quinacridone pigments. Each specimen is offered in only one crystal modification. Unsubstituted quinacridone is somewhat of an exception in that two crystal phases are commercially available.
Table 3.2 Commercially available quinacridone pigments.

|
||||||||||
| C.I. Name | C.I. Constitution Number | R2 | R3 | R4 | R9 | R10 | R11 | Range of shades | Total number of known modifications | |
| Quinacridone pigments | ||||||||||
| P.V.19 | 73900 | H | H | H | H | H | H | red-violet (β-phase), bluish red (γ-phase) | 4 | |
| P.R.122 | 73915 | CH3a) | H | H | CH3 | H | H | bluish red (magenta) | 4 | |
| P.R.192 | — | CH3 | H | H | H | H | H | bluish red | 1 [51] | |
| P.R.202 | 73907 | Cl | H | H | Cl | H | H | bluish red to violet | 1 (2,9-dichloro: 4) | |
| solution with unsubstituted and monochloroquinacridone | ||||||||||
| P.R.207 | 73906/ | H | H | Cl | H | H | Cl | yellowish red | 1 (4,11-dichloro:4) | |
| 73900 | solid solution with unsubstituted quinacridone | |||||||||
| P.R.209 | 73905 | H | Cl | H | H | Cl | H | red | 1 | |
| mixed with 1,8- and 1,10-dichloroquinacridone | ||||||||||
| P.R.282 | — | solid solution of 2,9-dimethylquinacridone with 2-methylquinacridone and unsubstituted quinacridone | magenta | — | ||||||
| Quinacridone quinone pigments (Quinacridone quinone, C.I. 73920) | ||||||||||
| P.R.206 | 73900/ | H | H | H | H | H | H | maroon (golden yellow) | 1 | |
| 73920 | solid solution with quinacridone quinone | |||||||||
| — | — | H | H | Cl | H | H | Cl | scarlet | 1 (4,11-dichloro:4) | |
| solid solution with quinacridone quinone | ||||||||||
| P.O.48 | — | solid solution of unsubstituted quinacridone + quinacridone quinone | maroon (golden yellow) | 1 | ||||||
| P.O.49 | — | Unsubstituted quinacridone | maroon (golden yellow) | 1 | ||||||
| + quinacridone quinone | ||||||||||
| P.V.42 | — | — | maroon | 1 | ||||||
| P.V.55 | — | solid solution of 2,9-dimethoxyquinacridone and 2,9-dichloroquinacridone | bluish violet | 1 | ||||||
| a Also available as a solid solution with unsubstituted quinacridone. | ||||||||||
3.2.4.2 Pigment Violet 19, β-Modification
The β-modification affords a reddish violet shade. These pigments are frequently used in combination with inorganic pigments, especially with iron oxide, or to a decreasing extent with Molybdate Red pigments, to colour industrial paints. Such pigment combinations provide comparatively dull shades of red to bordeaux. The excellent hiding power of the resulting systems is attributed to the content of highly scattering inorganic pigments with higher refractive indices. Combinations with opaque organic pigments, especially the P.O.36 type, are equally common. The products are used mostly in original automotive and automotive refinishes. The shade of the β-modification of P.V.19 is close to that of P.R.88, a tetrachlorothioindigo pigment. The tinctorial strength of the reddish violet quinacridone grades, however, is considerably higher in combination with these inorganic pigments, especially the Molybdate Red types. They also produce cleaner shades than thioindigo pigments. Quinacridone pigments, despite their higher price, have the advantage of being more economical in use. Moreover, the β-modification of P.V.19 is also more lightfast and weatherfast than its thioindigo counterparts. Thioindigo pigments and quinacridone pigments, however, demonstrate different metameric effects (also in combination with inorganic pigments) and may thus not be exchanged directly. The higher tinctorial strength of quinacridone pigments is less noticeable in white reductions, but the pigment is appreciably more lightfast and weatherfast. Full shades tend to darken. The β-modification of P.V.19, like other quinacridone pigments, shows good resistance to the common organic solvents. Incorporated in baked enamels, the pigment is correspondingly fast to overcoating. The systems show excellent fastness up to 140 °C, while some bleeding is observed at 160 °C. In this respect, the β-crystal modification of P.V.19 is inferior to the γ-form. Paints made of β-P.V.19 are fast to acid and alkali.
Like several other quinacridone pigments, P.V.19 types of the β-modification are prone to cause problems, especially through pigment flocculation, while being dispersed in paint systems.
The different grades exhibit a wide range of particle size distributions. As with other pigments, a decreasing average particle size is accompanied by enhanced transparency in full shade, increased tinctorial strength in white reduction and a general colour shift towards more intense reddish violet shades. There is no noticeable change in lightfastness and weatherfastness if the particle size is changed. The transparent types are also suited to application in metallic finishes. In addition, they lend colour to various other media throughout the paint industry wherever high lightfastness and weatherfastness, chemical fastness or heat stability are required. The list includes coil coating and various types of powder coatings, although it is recommended to adjust the pigment to the hardener before it is applied in a powder coating. β-P.V.19 does not show plate-out in most powder coatings. The β-modification of P.V.19 is also used as a colourant for plastics. In the colouration of PVC and PUR coatings, β-P.V.19 is also often used in combination with inorganic iron oxide and Molybdate Red pigments. The pigment is especially suitable for applications that require high hiding power as well as good lightfastness and weatherfastness, such as for automobile interiors, purses, and canvasses for tarps, awnings and so on. Importantly, β-P.V.19 is more lightfast and weatherfast in combination with Iron Oxide or Molybdate Red pigments than in combination with TiO2.
The β-modification of P.V.19 is very fast to bleeding in plasticized PVC. Its lightfastness in this medium satisfies the requirements for long-term weathering, as long as the demands are not exceptionally high. The pigment is thermally stable in polyolefins, it withstands exposure up to 300 °C. Its tinctorial strength in polyolefins, like in other media, is average. Approximately 0.2% pigment is needed to formulate 1/3 SD HDPE samples containing 1% TiO2, if a tinctorially strong type with a high specific surface area is used. However, the pigment noticeably affects the shrinkage of PE and other partially crystalline polymers. High heat stability makes β-P.V.19 a suitable colourant for polystyrene and other industrial synthetics. Likewise, β-P.V.19 is also used in spin dyeing, for instance of PP. The printing ink industry utilizes β-P.V.19 only where high lightfastness or other special fastness properties are required. It is found, for instance, in inks for printing on PVC and for posters.
Metal deco prints containing β-P.V.19 withstand up to 180 °C for 10 min and 170 °C for 30 min, respectively. The prints are fast to sterilization and calendering and to several special-purpose media, such as particular organic solvents.
3.2.4.3 Pigment Violet 19, γ-Modification
The γ-crystal modification of P.V.19 affords bluish red shades that are much yellower than those obtained by the β-modification. Commercial types of γ-P.V.19 show a wide range of particle size distributions. The specific surface areas vary accordingly; they range from approximately 30 to 70 m2 g−1.
The particle size of γ-P.V.19 has more of an effect on the lightfastness and weatherfastness of the resulting pigmented system than can be said of the β-modification. The grades of γ-P.V.19 with finer particle sizes, although less lightfast and weatherfast, have the advantage of being more transparent, tinctorially stronger and bluer than varieties with coarser particle sizes.
High lightfastness and durability makes γ-P.V.19 a useful product for various media throughout the coatings and paints industry, including high grade industrial paints and automotive finishes. γ-P.V.19 is used both in solid shades and as a shading pigment. Applied in combination with Molybdate Red, γ-P.V.19 affords comparatively brilliant, opaque shades, which are not accessible by using other quinacridone pigments. Moreover, γ-P.V.19 is also utilized in combination with other opaque pigments. Full shades and similarly deep shades darken slightly as they are exposed to light or weather. On the whole, γ-P.V.19 grades with fine grains are less weatherfast than the β-crystal modification and also various other types, such as P.R.122. The products of γ-P.V.19 with coarser particle sizes, however, frequently perform better than these quinacridone pigments. Excellent weatherfastness makes γ-P.V.19 a suitable candidate for exterior house paints. Its use in these areas, however, is somewhat limited by the fact that long-term exposure to alkali, for instance on freshly applied plaster, may considerably reduce the lightfastness and weatherfastness of the product as well as its tinctorial strength.
The stronger and more economical types with fine particle sizes are also found in transparent paints. If it is to be used in two-coat metallics, however, the application of the pigment must be combined with UV absorbants in topcoats to render it weatherfast enough to be applied in original automotive finishes.
The printing ink industry mostly prefers the type with finer particle sizes. These are incorporated into high grade printing inks for metal deco and poster printing. Combination with 2,9-dimethylquinacridone (P.R.122) affords a bluish shade of red, which closely approaches the standard magenta for three- and four-colour printing. Compared to other types of pigments covering the same portion of the spectrum, γ-P.V.19 types lack tinctorial strength. Inks formulated at almost 20% pigment, for instance, are needed to produce 1/3 SD letterpress proof prints at standard conditions. The γ-crystal modification is thus utilized primarily in areas where other pigments are not fast enough to satisfy the use requirements. In terms of lightfastness, 1/3 SD letterpress proof prints equal step 6–7 on the Blue Scale. The prints are thermally stable up to 190 °C for 10 min or to 170 °C for 30 min. They are fast to sterilization and calendering. Excellent fastness properties make γ-P.V.19 an equally suitable pigment for decorative printing inks for melamine and polyester based laminated plastic sheets. The samples equal step 8 on the Blue Scale for lightfastness.
γ-P.V.19 is frequently used for its high heat stability to replace more expensive bluish Cadmium Red pigments (cadmium selenide/cadmium sulfide mixed crystals) in lightfast thermoplastic systems; 0.2% pigment is required to formulate 1/3 SD HDPE samples containing 1% TiO2. Some types exhibit a very moderate tendency to nucleate, which is why there is no limitation to pigment use in HDPE.
γ-P.V.19 as a colourant for plastics is available both as a powder and as a pigment preparation containing predispersed pigment. The list of applications includes PVC films, technical articles and toys made of PVC or PO, as well as spin dyeing products of PP fibres and PP monofilaments. Employed in polyester spin dyeing, the pigment is heat stable enough to be used in the condensation process at 240–290 °C for 5–6 h. A certain colour change at low concentrations is attributed to the fact that γ-P.V.19 dissolves in polyester, a feature that makes it unsuitable as a shading pigment. Like in other media, the pigment is often incorporated in polymers together with grades of Molybdate Red.
The γ-form of P.V.19 is also applicable in injection-moulded and extrusion-made polyamide. It satisfies not only the high thermal requirements in connection with these purposes but has the added advantage of being, like P.R.122 and 209, chemically inert to the slightly alkaline and reducing plastic melt.
Moreover, γ-P.V.19 is also found in various other media, such as powder coatings and cast resins. This includes systems based on unsaturated polyester resins whose hardening is not affected by the pigment. The list of application media includes plastics that are processed at very high temperature (such as polycarbonates), in which the pigment is thermally stable up to 320 °C. PUR foams and polyacetals should also be mentioned, although γ-P.V.19, like other quinacridone pigments, tends to migrate in polyacetals if it is used at concentrations below 0.1%. The pigment is also found in watercolours for artists.
3.2.4.4 Pigment Red 122
2,9-Dimethylquinacridone is more weatherfast than most other unsubstituted types. It possesses excellent fastness to migration and outstanding heat stability. P.R.122 offers a very clean bluish shade of red, which is usually referred to as pink or magenta. Its main areas of application are in high grade paints, printing inks and plastics, which is also true for the γ-modification of unsubstituted quinacridone.
P.R.122, which is more durable than unsubstituted types of quinacridone with fine particle sizes, may safely be used in automotive metallic finishes. Highly transparent types are available for this important purpose. However, such finishes occasionally present problems in terms of viscosity and flow. Its clean bluish shade makes P.R.122 an equally suitable pigment for use in combination with Molybdate Orange. The resulting colourations are particularly lightfast and weatherfast in automotive finishes. The thus-prepared shades are distinctly cleaner than combinations of Molybdate Red with β-P.V.19. Combinations of Molybdate Red with the γ-modification are less economical than those with P.R.122 and afford somewhat cleaner and more bluish shades. The pigment is used primarily as a shading pigment. Excellent stability to common organic solvents renders the pigment completely fast to overcoating in oven drying systems up to 160 °C. In this respect, P.R.122 performs better than the β-modification of unsubstituted quinacridone. Moreover, P.R.122 is recommended for use in architectural paints: its excellent weatherfastness is of interest for exterior application. P.R.122, like P.V.19, is also used in powder coatings.
2,9-Dimethylquinacridone has a slightly nucleating effect on partially crystalline polymers such as HDPE. The plastics industry therefore uses P.R.122, like the γ-modification of unsubstituted quinacridone, primarily in systems such as PVC films and coatings made from PVC or from PUR, in technical products or toys made from PVC or polyolefins, or in PO films. In addition, P.R.122 is also utilized in PP spin dyeing. In polyester spin dyeing, P.R.122 satisfies the high thermal requirements of the condensation process (exposure to 280 °C for 5–6 h). At low concentrations, however, the pigment dissolves to a certain extent in its medium and accordingly changes colour. Its lightfastness is also affected. Plastics are advantageously coloured with pigment powders or preparations containing predispersed pigment. P.R.122 grades are considerably stronger than γ-P.V.19, they match the β-modification in tinctorial strength: 0.21% pigment is required to produce a 1/3 SD HDPE sample containing 1% TiO2. Comparative systems containing γ-P.V.19 are formulated at 0.26% pigment. P.R.122 demonstrates excellent heat stability, which makes it a suitable colourant for polystyrene, impact-resistant polystyrene, ABS and polycarbonate. In addition, it is also used in various other media wherever high lightfastness, good heat stability and other special fastnesses are a prime consideration. Examples are systems such as unsaturated polyester resins and PUR foams.
P.R.122, like other quinacridone pigments, shows excellent application properties in high grade printing inks. It is fast to sterilization and to calendering. However, P.R.122 is somewhat weaker in these media than P.V.19 types. At standard film thickness, 1/3 SD letterpress proof prints are made from printing inks that contain approximately 25% pigment. To compare values, notably, approximately 13% are needed of the β-modification of P.V.19 and about 19% of the γ-modification. The standard magenta for three- and four-colour printing on cans, posters, and packaging may be approached by combining P.R.122 with pigments of the γ-P.V.19 type. P.R.122 is also found in decorative printing inks for laminated plastic sheets. Complete fastness to the solvents that are commonly used in this area, such as styrene monomer and acetone, makes P.R.122 a useful colourant for polyester-based sheets, as well as for melamine resin systems. The lightfastness of such products equals step 8 on the Blue Scale.
3.2.4.5 Pigment Red 192
Production of P.R.192, 2-methyl-quinacridone, has been discontinued. The pigment never gained considerable commercial recognition. It provides a shade somewhere between the shades of the quinacridone pigments P.R.122 and γ-P.V.19. The commercial type was distinguished by good transparency, but inadequate flow behaviour. In some systems, the pigment was almost as durable as P.R.122. It was recommended for use in high grade industrial paints, especially for automotive finishes, plastics and special-purpose printing inks.
3.2.4.6 Pigment Red 202
P.R.202, a very lightfast and weatherfast solid-solution quinacridone pigment, provides more bluish and considerably duller shades than 2,9-dimethylquinacridone. (2,9-Dichloroquinacridone, which is listed in the Colour Index as Pigment Violet 30, has been discontinued.) P.R.202 is primarily applied in automotive finishes. Even highly transparent types with fine particle sizes provide two-coat metallic finishes that offer excellent rheological properties. P.R.202 is accordingly superior to 2,9-dimethylquinacridone (P.R.122). On the other hand, metallic finishes made from P.R.202 are duller and more bluish. P.R.202 types exhibit more apparent flop (Section 3.1.5.3) than grades of P.R.122. At similar transparency, the solid-solution pigment is more durable than the individual components, especially more so than unsubstituted quinacridone pigment. Its response to weathering corresponds to that of 2,9-dimethylquinacridone. Two-coat finishes made of P.R.202, however, are frequently less weatherfast than single-layer products. In terms of fastness to overcoating and other aspects of pigment performance and properties, P.R.202 largely behaves like P.R.122.
A version of P.R.202 in a platelet form is obtainable by recrystallizing of the crude pigment from a polar solvent such as DMF in the presence of a thiol compound and sodium methoxide. The new form affords, for instance in PVC, the appearance of a lustrous copper bronze tone [92].
3.2.4.7 Pigment Red 207
P.R.207 is a pigment made from a solid solution of unsubstituted and 4,11-substituted dichloroquinacridone in a ratio of about 2 : 1. Pure 4,11-dichloroquinacridone is not commercially available. P.R.207 affords a yellowish red shade, which is very yellowish compared to other non-oxidized quinacridone pigments. P.R.207 is still somewhat yellower, but also duller than P.R.209. The commercial type of P.R.207 is very opaque. In fact, it has better hiding power and is considerably weaker in white reductions than the commercially available version of 3,10-dichloroquinacridone. P.R.207 demonstrates excellent weatherfastness, and its response to weathering approximately equals that of P.R.209. This is also true for other aspects of pigment performance, including resistance to organic solvents, fastness to overcoating, tendency to flocculate and other properties and fastnesses. P.R.207 is used primarily in automotive finishes but is also utilized as a colourant for plastics and artists' colours.
3.2.4.8 Pigment Red 209
P.R.209 is a speciality product. It is used in much less volume than the β- and γ-modifications of unsubstituted quinacridone and also than 2,9-dimethylquinacridone. P.R.209 has stimulated a certain amount of interest, not only for its excellent lightfastness and weatherfastness, migration fastness and heat stability, which are comparable to those of P.R.122, but also for its very yellowish shade of red, which is unusual for a quinacridone pigment.
P.R.209 is applied in paints, especially in various types of automotive finishes. Exceptionally clean shades of red are furnished if P.R.209 is combined with Molybdate Orange. There is a certain disadvantage to the fact that such combinations are formulated at a relatively high P.R.209 content. The need for a high pigment content considerably compromises the economy of such systems in contrast to pigment combinations with other quinacridones. Moreover, P.R.209 is found in metallic automotive finishes, for instance in conjunction with transparent Iron Oxide pigments. Comparatively poor tinctorial strength, however, restricts the use of P.R.209 in this area to occasions where the desired shade is not accessible through pigments derived from 2,9-dimethylquinacridone or 2,9-dichloroquinacridone.
Other areas of application include several plastics that are processed at elevated temperature. P.R.209 lends colour to polyolefins (in which it only slightly affects the shrinkage), to ABS, to polyacetal resins and other industrial polymers.
3.2.4.9 Quinacridone Quinone Pigments
In contrast to most quinacridone pigments, commercial products based on quinacridone quinone afford relatively dull, slightly less weatherfast red and reddish to yellowish shades of orange. These pigments are used as special-purpose products to create metallic effects in paints. It is possible, particularly in combination with transparent Iron Oxide Yellow pigments, to produce interesting shades of gold for fashionable automotive finishes, but these products exhibit more of a flop than transparent grades of quinacridone pigments. These solid-solution pigments are used particularly throughout the USA.
3.2.4.9.1 Pigment Red 206
P.R.206 is a solid-solution and consists of unsubstituted quinacridone and quinacridone quinone in a ratio of about 2 : 1 in the lattice of quinacridone quinone [71]. P.R.206 affords a very dull, yellowish shade of red, referred to as maroon. The pigment is considerably weaker than perylene pigments. All commercially available types of P.R.206 are more or less transparent and are used mostly in metallic finishes for automobiles, to which they lend reddish shades of copper. The pigment is often found to be difficult to disperse. The finishes frequently exhibit rheological problems, especially at high pigment concentration.
3.2.4.9.2 Pigment Orange 48
P.O.48, like P.R.206, is a solid solution of unsubstituted quinacridone (C.I. 73900) and quinacridone quinone (C.I. 73920) in a ratio of about 70 : 30 [71]. The full shade is a deep maroon, which turns into a yellowish brown as TiO2 is added, such as in 1 : 5 TiO2 reductions. The main area of application for P.O.48 is in metallic finishes, to which the pigment lends interesting shades of copper and gold. P.O.48 produces considerably redder shades than P.O.49, a member of the same class of pigments. P.O.60, a benzimidazolone pigment, affords colours that at equal conditions are intermediate between those of the two quinacridone quinone pigments. Newly developed grades of P.O.48 and 49 have been introduced offering appreciably higher weatherfastness, which now corresponds to that of other representatives of the same series that are found in original automotive finishes. Moreover, these novel types also exhibit improved flow behaviour. P.O.48 is also found in plastics and is recommended for spin dyeing. It dissolves in polyamide and in various other polymers.
3.2.4.9.3 Pigment Orange 49
The exact stoichiometry of this quinacridone/quinacridone quinone pigment remains to be published. P.O.49, like P.R.209, is a speciality product for metallic shades. It is used to produce shades of gold in finishes, which are considerably more yellowish than those of P.O.48.
3.2.4.9.4 Pigment Violet 42
The chemical constitution of P.V.42, a solid-solution pigment, remains to be published. P.V.42 has little commercial value. Its shade is a maroon, which is used primarily to afford metallic shades in original automotive finishes.
3.2.4.9.5 Pigment Violet 55
A new solid solution pigment has been described, which is composed of the blue-violet shade of 2,9-dimethoxyquinacridone as guest in the very durable 2,9-dichloroquinacridone (P.R.202) host crystal lattice. Generally solid-solution technology may give significantly better properties (fastness, hue stability) than can be achieved from single blending [93].
3.2.4.10 Other Quinacridone Pigments
Several other members of this class are also available, although not all are listed in the Colour Index. Their exact chemical and physical properties remain to be published. The list includes solid-solution pigments made of unsubstituted quinacridone with dimethylquinacridone or other substituted quinacridones. Solid-solution pigments of two substituted quinacridones have been published, too [94]. These pigments are considered speciality products for automotive finishes, especially for metallic finishes.
3.3 Vat Dyes Prepared as Pigments
In Sections 3.4 3.6, and 3.7 several polycyclic pigments are discussed that have been used for a long time as vat dyes for textile fibres. Heading the list are perylene, perinone and thioindigo pigments, as well as pigments derived from anthraquinone.
Original attempts at using these vat dyes as pigments afforded only dull shades with insufficient tinctorial strength. As pigment technology developed into a well-established industry and chemical and physical methods were found to increase the quality of pigments, vat dyes stimulated more and more interest throughout the pigment industry. To employ a vat dye as a pigment, it was necessary to improve the chemical purity of the product, to optimize the particle size and the particle size distribution, and to synthesize certain target crystal modifications.
The polycyclic colourants discussed in this context (and this is also true for copper phthalocyanine and quinacridone pigments) as a rule evolve from the manufacturing process as products with relatively coarse particles, which is in contrast to hydrazone pigments (Section 2.2.3).
Although a crude pigment may be converted into a material with finer particle sizes by milling, there is a certain disadvantage to the fact that the frequently wide particle size distributions (approx. 0.5–200 µm) make these compounds unsuitable for use as pigments. The crystallinity and deagglomeration is commonly improved by treating the pigment in an organic solvent or water in the presence of a dispersing agent. This type of aftertreatment is referred to as finishing a pigment.
Various methods of aftertreatment have been developed to transform vat dyes into easily dispersible pigments. Heading the list are the following methods:
- reduction to form a leuco base, removal of impurities, and subsequent reoxidation;
- dissolving the crude product, for instance in sulfuric acid, precipitation in water and/or organic solvents, possibly in the presence of surfactants at exactly defined conditions;
- formation of a pigment sulfate in the presence of 70–100% sulfuric acid, isolation of the intermediate, and subsequent conversion into the pure pigment by treatment with water, possibly in the presence of surfactants, and heating;
- thermal aftertreatment in water and/or organic solvents;
- through various dispersion and milling techniques – particle size reduction may be achieved, for instance, through fine dispersion in kneaders, fast rotating stirrers with milling effect, or in grinding units based on the impact principle; some crush the material through mutual friction between particles or by rotation or vibration; vibration mills, roll mills or ball mills may be used; the crude pigment is usually prepurified, for instance by reprecipitation, before being dispersed;
- milling the pigment in the presence of salts or solvents, possibly in the presence of surfactants, or in the presence of strong, non-oxidizing acids (with a pKα below 2.5).
Often two or more of these processes are combined as necessary to optimize the results.
The methods that are used to convert individual vat dyes into pigments are described in the respective sections.
Despite the extensive technology that has developed in this field, very few of the originally large number of useful vat dyes continue to play a role as pigments. The somewhat narrow selection is also made on the basis of the cost/performance considerations for each of these relatively complicated compounds. Very few of these synthetically demanding pigments match the fastness standards of phthalocyanine pigments.
3.4 Perylene and Perinone Pigments
Perylene and perinone pigments are chemically related. The group of perylene pigments is derived from perylene-3,4,9,10-tetracarboxylic acid (48), while perinone pigments are derivatives of naphthalene-1,4,5,8-tetracarboxylic acid (49), and semiperinone pigments are based on naphthalene-1,8-dicarboxylic acid (50):
Members of both groups are manufactured by essentially the same route. Starting from the anhydrides of 48, 49 and 50, formation of bisimido or, respectively, bisimidazole compounds is accomplished with amines or diamines. The compounds have been known as vat dyes since 1913 (perylenes) and 1924 (perinones).
3.4.1 Perylene Pigments
Perylene pigments are diimides of perylene tetracarboxylic acid. The unsubstituted compound, although never used as a vat dye, is the oldest known member of this class. It was described as early as 1912. In due course, several perylene tetracarboxylic acid diimides emerged (dimethylimide in 1913), which were initially used exclusively as vat dyes. It was not until 1950 that perylene compounds found use as pigments. This shift in emphasis is credited to investigations by Harmon Colours, which broke new ground in describing the conversion of vat dyes into pigments. Several members of the perylene pigment series have thus found their way towards industrial-scale production.
The customary method of preparing perylene pigments is by reaction of perylene tetracarboxylic dianhydride with primary aliphatic or aromatic amines in a high boiling solvent. The dianhydride itself is also used as a pigment. Dimethylperylimide may also be obtained by treating the diimide with methyl chloride or dimethyl sulfate.
3.4.1.1 Preparation of the Starting Materials
The primary starting material for the synthesis of perylene tetracarboxylic acid pigments is the dianhydride 48a. It is prepared by fusing 1,8-naphthalene dicarboxylic acid imide (naphthalic acid imide 51) with caustic alkali, for instance in sodium hydroxide/potassium hydroxide/sodium acetate at 190–220 °C, followed by air oxidation of the molten reaction mixture or of the aqueous hydrolysate. The reaction initially affords the bisimide (peryldiimide) 52, which is subsequently hydrolysed with concentrated sulfuric acid at 220 °C to form the dianhydride:

A new imaging method executed by scanning tunnelling microscopy has shown clearly the molecular structure of perylenetetracarboxylic acid anhydride (Figure 3.22) [95].


Figure 3.22 Scanning electron microscopic image of perylene tetracarboxylic acid anhydrate [95].
Naphthalic acid imide (50) is obtained through air oxidation of acenaphthene (53) with vanadium peroxide as a catalyst. The intermediate, naphthalic anhydride (54), is subsequently reacted with ammonia:

3.4.1.2 Chemistry, Manufacture
Perylene pigments are derivatives of the general structure 55:

In industrially interesting pigments, X usually stands for O or NR, and R represents H, CH3 or possibly a substituted phenyl moiety. The phenyl ring may possess a methyl, a methoxy, an ethoxy, or a substituent:

Heating perylene tetracarboxylic dianhydride 52, as described in Section 3.4.1.1, to 150–250 °C with a primary aliphatic or aromatic amine in a high-boiling solvent affords the desired crude pigment. The reaction may be accelerated by adding agents such as sulfuric acid, phosphoric acid or zinc salts [96]. Pigment synthesis through condensation in aqueous medium has also been described.
A different route proceeds by alkali fusion of the correspondingly substituted naphthalic acid imide. This pathway parallels the synthesis of perylene tetracarboxylic acid diimide 51 (Section 3.4.1.1). The method is particularly suited to aliphatic amines.
Unsymmetrically substituted perylene pigments are a comparatively recent novelty. Selective protonation of the tetra sodium salt of perylene tetracarboxylic acid affords the monosodium salt of perylene tetracarboxylic monoanhydride in high yield. Stepwise reaction with amines produces unsymmetrically substituted perylene pigments [97].
There are various techniques of aftertreating the crude product which convert it into an industrially useful pigment. Important methods include reprecipitation from sulfuric acid, milling, and recrystallization from solvents. It is also customary to combine these methods in order to optimize the results [98].
The dianhydride of perylene tetracarboxylic acid is converted into the pigment form by preparing the corresponding alkali salt and then reprecipitating the compound with an acid. The dianhydride is formed after separating the acid by thermal aftertreatment at 100–200 °C, possibly under pressure, with an organic solvent. The list of suitable media includes alcohols, ketones, carboxylic acid esters, hydrocarbons and dipolar aprotic solvents.
Opaque perylene pigment varieties are treated in ball mills, either in the presence or absence of milling auxiliaries. The pigments are typically heated to 80–150 °C in solvents such as methyl ethyl ketone, isobutanol, diethylene glycol, and N-methylpyrrolidone, possibly in the presence of water.
Perylene tetracarboxylic acid dimethylimide may also be prepared by methylating the corresponding diimide.
3.4.1.3 Crystal Structures and Properties
Perylene pigments exist in a wide range of hues, they provide red, bordeaux, violet, brown and black shades. The pigments exhibit excellent solvent stability, good-to-very good migration stability in plastics, fastness to overcoating in paints, high chemical inertness and superior thermal stability. Only the basic compound, the dianhydride of perylene tetracarboxylic acid, is not fast to alkali.
As a group, perylene pigments offer high tinctorial strength. They are frequently found to be appreciably stronger than quinacridone pigments. Moreover, perylene pigments provide excellent lightfastness and weatherfastness. In this respect, they approximately equal the performance of quinacridone pigments.
Several perylene pigments are polymorphic, including P.R.224 and P.R.149. Crystal structures have been determined for a large series of commercial and non-commercial perylene pigments by single-crystal X-ray diffraction [99–105], by X-ray powder diffraction [106] and by electron diffraction [107]. The perylimide moiety is always almost planar. Alkyl and aryl substituents protrude from this plane. Phenyl groups attached to the imide nitrogen atoms are rotated by almost 90° from the perylene plane (Figure 3.23).

Figure 3.23 Molecular structure of P.R.123 (R = p-ethoxyphenyl) in the solid state [99]. View exactly perpendicular to the perylene moiety.
P.Bl.32, with p-methoxybenzyl substituents, adopts an S-shaped geometry (Figure 3.24).

Figure 3.24 Molecular structure of P.Bl.32 (R = p-methoxybenzyl) in the solid state [100].
In all perylene pigments, the molecules adopt a very dense packing in the crystals (Figures 3.25–3.33). The density reaches a value of 1.67 g·cm−3 for P.R.224, which is an extremely high density for an organic compound containing C, H, N and O atoms only. The mean atomic volume for organic crystals, which is generally around 18 Å3 per atom (H atoms not counted) [108], drops to 13.0 Å3 per atom for P.R.224 (Figures 3.25 and 3.26). The molecules are held together by strong van der Waals interactions, supported by some Coulomb interactions between the partially charged atoms. Hydrogen bonds are only present in P.V.29 (X = NH) (Figure 3.27) and in a few non-commercial compounds containing, for example, hydroxyalkyl substituents.

Figure 3.25 Crystal structure of P.R.224: (a) α-phase [101] and (b) β-phase [102].

Figure 3.26 The almost perfect space filling in a molecular layer of the α-phase of P.R.224.

Figure 3.27 Crystal structure of P.V.29 [103].
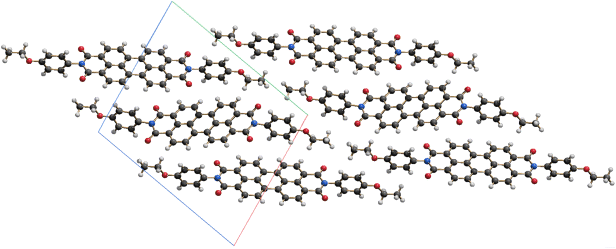
Figure 3.28 Crystal structure of P.R.123. The unit cell contains two symmetrically independent molecules.

Figure 3.29 Crystal structure of P.R.149 (α-phase) [104]. The unit cell contains two symmetrically independent molecules. The xylyl groups are drawn with reduced ball sizes to enhance clarity.

Figure 3.30 Crystal structure of P.R.178 [99].

Figure 3.31 Crystal structure of P.R.179 [100].

Figure 3.32 Crystal structure of P.Bl.31 [100].

Figure 3.33 Crystal structure of P.Bl.32.
Interestingly, the colours of perylene pigments vary from orange (R = ethyl) via scarlet–red (R = p-ethoxyphenyl), red (R = xylyl) (Figures 3.36 and 3.37), bluish red (R = p-methoxyphenyl) to black (p-methoxybenzyl), although the substituent R does not take part in the chromophoric perylene system (Table 3.3). The reason for this has been sought in exciton interactions between neighbouring molecules [109]. Depending on the steric requirement of the substituent R, the crystal structure varies, causing variations in the relative position and orientation of the neighbouring molecules (Figures 3.34–3.39), leading to differences in the exciton interactions and thus modifying the absorption bands and, hence, changing the colour. In 1989 Klebe et al. found an empirical correlation between the relative position of neighbouring molecules and λmax [99].
Table 3.3 Optical properties of some perylene pigments [99].
| R | Figure | λmax (nm) | Colour | C.I. Name |
| C2H5 | 3.34 | 500 | orange | — |
| PhOC2H5 | 3.35 | 554 | scarlet to red | P.R.123 |
| PhNNPh | 3.36 | 565 | red | P.R.178 |
| CH3 | 3.37 | 569 | red to maroon | P.R.179 |
| CH2CH2Ph | 3.38 | 628 | black | P.Bl.31 |
| CH2PhOCH3 | 3.39 | 678 | black | P.Bl.32 |

Figure 3.34 Arrangement of neighbouring molecules in an orange non-commercial perylene pigment (R = ethyl) [100]. View exactly perpendicular to the perylene moiety. The crystal contains two symmetrically independent molecules with slightly different neighbourhoods.

Figure 3.35 Arrangement of neighbouring molecules in P.R.123. The crystal contains two symmetrically independent molecules.
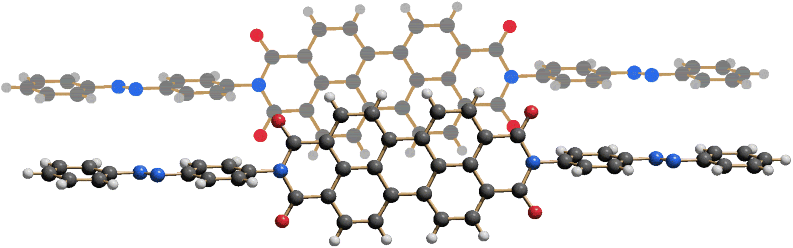
Figure 3.36 Arrangement of neighbouring molecules in P.R.178.

Figure 3.37 Arrangement of neighbouring molecules in P.R.179.

Figure 3.38 Arrangement of neighbouring molecules in P.Bl.31. The terminal groups are drawn with reduced ball sizes to enhance clarity.

Figure 3.39 Arrangement of neighbouring molecules in P.Bl.32. The terminal groups are drawn with reduced ball sizes to enhance clarity.
3.4.1.4 Application
Most perylene pigments, like quinacridone pigments, are used primarily in high grade industrial paints, especially in original automotive finishes and in automotive refinishes. Different types are available of some pigments, ranging from varieties with very fine particle sizes and high specific surface areas and correspondingly high transparency to versions with coarse particle sizes and low specific surface areas as well as high hiding power. Types with fine particle sizes are used especially in metallic and transparent paints, while opaque types provide full shades, frequently in combination with inorganic or other organic pigments. Some types are particularly suited to use in plastics and in spin dyeing products, in which they demonstrate excellent heat stability. However, perylene pigments are rarely used to colour HALS (hindered amine light stabilizer) stabilized polyolefins, that is, polyolefins that are stabilized by steric amines. At medium to high pigment concentrations, these stabilizers may be inactivated or even destroyed by exposure to light, rapidly converting the polyolefin system into a brittle material.
3.4.1.5 Commercially Available Perylene Pigments
3.4.1.5.1 General
Table 3.4 lists representative commercial perylene pigments.
Table 3.4 Commercially available perylene pigments.

|
|||
| CI. Name | C.I. Constitution Number | X | Shade |
| RR.123 | 71145 |  |
scarlet to red |
| P.R.149 | 71137 |  |
red |
| P.R.178 | 71155 |  |
red |
| P.R.179 | 71130 | H3CN | red to maroon |
| P.R.190 | 71140 |  |
bluish red |
| P.R.224 | 71127 | O | bluish red |
| P.V.29 | 71129 | HN | red to bordeaux |
| P.Bl.31 | 71132 |  |
black |
| P.Bl.32 | 71133 |  |
black |
3.4.1.5.2 Pigment Red 123
P.R.123 is not produced in large volume, although it has stimulated more commercial interest than the chemically closely related P.R.190. P.R.123 is primarily used in paints. The pigment provides a medium red shade of much higher cleanness than that of P.R.190. P.R.123 is somewhat duller and slightly yellower than P.R.178. Transparent types are employed especially in shading systems for emulsion paints. P.R.123 offers high weatherfastness. In full shades and similarly deep shades, most grades darken slightly upon exposure to weather. In concentration ranges where high weatherfastness is a primary concern, P.R.123 is replaced by opaque types of the more durable P.R.178. Opaque varieties of P.R.123 are commonly used in combination with other organic or inorganic pigments, including TiO2, to suppress or cover up this darkening.
P.R.123 shows good resistance to organic solvents. At common baking temperatures, however, oven drying systems containing P.R.123 are not entirely fast to overcoating. Opaque versions of P.R.123 are used not only in industrial paints, including automotive finishes, but also in emulsion paints. Good weatherfastness makes these grades suitable colourants for exterior house paints. P.R.123 is alkali proof.
In plastics, P.R.123 retains its colour value up to 220 °C. At higher temperature, the colour changes towards bluer and duller shades. No further change is observed, however, between 240 and 300 °C. Incorporated in polyolefins, P.R.123 considerably affects the shrinkage of injection-moulded products, which excludes it from being recommended for use in these media. Although not entirely fast to bleeding in plasticized PVC, P.R.123 is used to a certain extent in PVC coatings. Plasticized PVC systems, like other media containing P.R.123, perform much more poorly than systems containing the somewhat yellower and considerably stronger, more heat-stable and more lightfast P.R.149, which is a member of the same class of pigments. P.R.123 is also used in spin dyeing. The pigment is even fast to the considerable heat that accompanies the condensation process of polyester spin dyeing (Section 1.8.3.9). In addition, P.R.123 is also used to colour PUR foam.
3.4.1.5.3 Pigment Red 149
P.R.149, a clean, medium to slightly bluish red pigment, is used primarily in the colouration of plastics. With a melting point above 450 °C, the pigment is exceptionally heat stable. Polyolefin systems containing P.R.149 may be processed up to 300 °C. Like other members of its class, P.R.149 affects the shrinkage of injection-moulded articles. Its influence, however, decreases with increasing temperature. P.R.149 exhibits high tinctorial strength. Less than 0.15% pigment is needed to formulate 1/3 SD HDPE systems containing 1% TiO2. For comparison, approximately 20% more of the somewhat bluer P.R.123 are required for the same purpose, although the two pigments have the same average particle size. Like other members of its class, P.R.123 destroys any HALS stabilizers present in plastics by exposure to light. Good heat stability makes the pigment a suitable colourant for polystyrene, impact-resistant polystyrene, ABS and other plastics that are processed at high temperature. P.R.149, especially in white reductions with TiO2, is much more heat stable than most quinacridone pigments.
In plasticized PVC, P.R.149 is very fast to migration. It also shows excellent tinctorial strength. 1/3 SD samples in plasticized PVC containing 5% TiO2 are formulated at only 0.63% pigment. Comparative values are: 0.75% P.R.123, 1.2% of the γ-modification of P.V.19, a quinacridone pigment, and 1.0% of P.R.122.
P.R.149 furnishes high lightfastness. Transparent colourations at 1/25 SD and opaque colourations at 1/3 to 1/25 SD equal step 8 on the Blue Scale for lightfastness. The pigment is not durable enough to be used in long-term exposure. The commercially available products are extremely transparent, a feature that is exploited in various applications.
P.R.149 also lends colour to cast resins made from materials such as unsaturated polyester or methacrylic acid methyl ester, which are polymerized with peroxide catalysts. P.R.149 is equally lightfast in these media. In polycarbonate, the pigment tolerates exposure to more than 320 °C. This is an asset in view of the fact that polycarbonate shows high melt viscosity and is thus processed at up to 340 °C. The list of applications also includes other media, such as PUR foams and elastomers, for which P.R.149 is recommended because of its good heat stability and its colouristic properties.
In addition, P.R.149 is also used to a considerable extent in the spin dyeing of polyacrylonitrile and polypropylene. Its high lightfastness presents an advantage in these media. In the concentration range between 0.1% and 3%, P.R.149 equals step 7 to step 7–8 on the Blue Scale. High pigment concentrations are to be avoided, however, in systems containing HALS stabilizers. P.R.149 performs perfectly with regard to the more important textile fastnesses, such as fastness to perspiration, dry and wet crocking, and resistance to perchloroethylene and similar solvents. It is often found in polyamide. Partial reduction of the pigment can lead to a change in shade towards a dull brown colour. On the fibre, the original colour of the pigment is quickly restored through oxidation. P.R.149 is an equally interesting candidate for the spin dyeing of polyester, although it does dissolve at low concentrations and consequently undergoes a colour shift towards orange. High thermal stability makes it resistant to the extreme temperatures to which it is exposed during the condensation process (Section 1.8.3.8).
The list of media that are coloured with P.R.149 includes not only polymers but also paints and printing inks, but only to a lesser degree. The pigment is used, for instance, in industrial finishes, where its high transparency is particularly useful in creating transparent and metallic effects in coatings. Not only is P.R.149 a pigment with high tinctorial strength, but it also offers high cleanness of shade and average lightfastness and weatherfastness. Full shades darken upon exposure to weather or light. In baked enamels, P.R.149 is not perfectly fast to overcoating. Special-purpose printing inks are available that are designed for substrates such as PVC films and metal deco prints. Such inks contain P.R.149 because of its fastness to organic solvents and the packaged products, as well as for its high heat stability (up to 220 °C for 10 min). Fastness to sterilization is another asset in this type of application.
3.4.1.5.4 Pigment Red 178
P.R.178 has been used for some time throughout the paint industry, where it is found especially in industrial finishes, including various types of automotive finishes. Two crystal modifications are known, only one of which possesses some commercial value. Only one version, a highly opaque type, is available. Full shades and similarly deep shades provide good lightfastness and weatherfastness, although some darkening is observed as the samples are exposed. The weatherfastness deteriorates rapidly in systems that are reduced with TiO2. P.R.178 covers the same range of shades as P.R.123, the shades are only slightly yellower and somewhat duller. Full shades of P.R.178 are more weatherfast than those of P.R.123, but the reverse is true for white reductions. P.R.178 offers good stability to the solvents commonly found in oven drying systems. In this respect, it performs approximately like other members of its class. The pigment is not completely fast to overcoating, but is thermally stable in excess of 200 °C.
As a colourant for plastics, P.R.178 exhibits good heat stability. The pigment shows an exceptionally strong effect on the shrinkage of injection-moulded polyolefin articles. P.R.178 is less lightfast than the traditionally used, somewhat yellower and noticeably stronger P.R.149. Like other perylene tetracarboxylic acid pigments, P.R.178 is not recommended for use in combination with HALS stabilizers. Its fastness to bleeding in plasticized PVC is very good and is equal to that of P.R.149. The commercial type of P.R.178 provides very opaque colourations, which are not accessible with other pigments covering the same portion of the spectrum.
According to the FDA Inventory of Effective Food Contact Substances P.R.178 can be used to colour food-contact articles.
3.4.1.5.5 Pigment Red 179
P.R.179, a dimethylperylimide compound, is probably the most significant member of its class. The pigment is primarily used in industrial coatings, especially for high grade original automotive (OEM) finishes and automotive refinishes. Colouristically, the pigment covers two different portions of the spectrum. On the one hand, it provides clean shades of red, with newer types producing comparatively yellowish shades. On the other hand, the pigment also affords maroon and bordeaux shades. Transparent types are available for metallic finishes, sometimes also in combination with other organic or inorganic pigments. These products extend the range of shades provided by quinacridone pigments towards the yellowish portion of the spectrum.
P.R.179 types demonstrate excellent weatherfastness. They are as weatherfast or even more weatherfast than substituted quinacridone pigments. The various types differ considerably in their flop behaviour. Some of the commercially available types provide high hiding power and are used especially in combination with Molybdate Red pigments to produce opaque dark red shades of high gloss and excellent weatherfastness. In paints, the pigments are heat stable up to 180 °C, sometimes even up to 200 °C. They demonstrate excellent fastness towards the organic solvents typically found in baking enamels. The systems are accordingly fast to overcoating. P.R.179 types, in contrast to those of P.R.224, are entirely fast to alkali. Dispersibility, fastness to rub-out and especially resistance to flocculation are frequently less than perfect. A new pigment grade for water-reducible automotive OEM finishes and refinishes is available on the market.
Although they are used to a considerable extent in paints, P.R.179 types have stimulated little interest throughout the plastics industry. The pigments are used in PVC and in PVC plastisols. Besides, P.R.179 is heat stable enough to be recommended for use in polyolefins. Like P.R.149 and other members of this class, P.R.179 undergoes considerable colour change if it is used in spin dyeing polyamide. This effect is attributed to the reducing effect of the melt. On the fibre, however, the pigment rapidly returns to its original colour.
3.4.1.5.6 Pigment Red 190
P.R.190 has a relatively small impact on the market. It is a speciality product for industrial paints, especially for automotive finishes, but offers no advantage over other members of its class. Its shade is dull and referred to as scarlet. In white reductions, the commercial type is very bluish and equally dull. The particle sizes of this product are too coarse for it to be used in metallic finishes. P.R.190 is very durable and very fast to organic solvents and to migration. The pigment is heat stable up to processing temperatures of 200 °C. Despite its good heat stability, the pigment possesses only limited commercial value as a colourant for plastics.
3.4.1.5.7 Pigment Red 224
Commercial types of perylene tetracarboxylic dianhydride offer a wide range of colouristic properties in application. The varieties range from highly transparent to very opaque grades. Both versions are used primarily in industrial paints, especially for automotive finishes. The transparent types are particularly interesting for metallic finishes. Since its chemical constitution renders the pigment sensitive to alkali, it fails to satisfy the stringent demands of the standardized test (Section 1.6.2.2). Other tests show that P.R.224 is not suited to single-layer metallic finishes. It is thus used primarily in base coat/clear coat metallic systems. The pigmented base coat, in Europe commonly a polyester lacquer, is protected against alkali, such as in car wash detergents, by a clear acrylic resin based varnish.
Commercial types exhibit a surprisingly wide range of shades. A new version has been introduced recently which, although performing like other versions of P.R.224 in terms of weatherfastness, solvent fastness and fastness to overcoating, is substantially yellower and noticeably cleaner than traditional types. Opaque varieties also provide brilliant shades of red. P.R.224 is used primarily in full shades, sometimes in combination with other organic or inorganic red pigments, such as Molybdate Red.
In addition, P.R.224 types are found in several other high grade industrial finishes. The pigments exhibit high tinctorial strength, which makes them suitable shading components for inorganic pigments. The resulting brilliant shades of red are considerably more yellowish than those furnished by quinacridone pigments. Because of the risk of chemical interaction, the pigment is not recommended for use in amine hardening binders. The same is true for exterior house paints that are applied onto stucco plaster. However, P.R.224 is very fast to organic solvents and overcoating. It is thermally stable up to 200 °C.
Apart from paints, P.R.224 is also used in polyacrylonitrile spin dyeing. Application in the spin dyeing of polypropylene is compromised by the fact that medium to high pigment concentrations accelerate the degradative action of light on HALS stabilizers (Section 3.4.1.4).
3.4.1.5.8 Pigment Violet 29
P.V.29 types demonstrate excellent weatherfastness, much more so than other perylene pigments. Their impact on the market, however, is limited by their very dull shade of maroon. Full shades are deep brown, almost black. The pigment is very fast to organic solvents and overcoating. Commercial types are utilized especially in metallic finishes. Full shades frequently bronze upon exposure to weather.
P.V.29 is also highly heat stable, which makes it a suitable colourant for plastics that are processed at high temperature. However, this advantage is somewhat compromised by its lack of cleanness.
P.V.29 is also used in polyester spin dyeing. It meets the high thermal demands of the condensation process, during which the pigment is exposed to 290 °C for 5–6 h. At 1/3 and 1/9 SD, the systems equal step 7 to step 8 on the Blue Scale for lightfastness. P.V.29 performs excellently in terms of the more important textile fastnesses, such as fastness to wet and dry crocking.
3.4.1.5.9 Various Other Perylene Tetracarboxylic Acid Pigments
Some other pigments of this class are found on the market. In full shade, these pigments provide tones of black or very dark olive or violet hues, which satisfy certain spectral requirements in the IR region. Examples are Pigment Black 31, C.I. 71132 and 32, C.I. 71133 (see Table 3.4). Their properties include resistance to certain chemicals and good weatherfastness.
3.4.2 Perinone Pigments
Perinones were first synthesized by Wilhelm Eckert and Heinrich Greune in Höchst a.M. in 1924 [110]. The inventors noticed their surprise to obtain coloured products, although the imines of naphthalene-tetracarboxylic acid are colourless. The production of perinones started already two years later, in 1926 [111]. The product showed a scarlet hue and consisted of a mixture of cis- and trans-isomers. It was sold under the brand name “Indanthrenscharlach 2G” [111], since 1929 as “Indanthrenscharlach GG” [112], and was used as a dye for the colouration of cotton and rayon, and for printing on cotton. In 1929/1930, the separation of the cis- and trans-isomers was achieved by fractional crystallization from sulfuric acid [113] or from a KOH/ethanol mixture [114]. Industrially, the KOH/ethanol route was used [115].
The clean orange trans-isomer was sold as Indanthrenbrillantorange GR [116], whereas the dull red cis-isomer had no commercial importance and was distributed as Indanthrenbordo 2R [116] or Indanthrenbordo RR [117]. Both isomers were used for a long time exclusively as vat dyes for cotton. It was not until 1950 that these compounds found recognition as pigments.
3.4.2.1 Preparation of the Starting Materials
Perinone pigments are obtained from naphthalene-1,4,5,8-tetracarboxylic acid or its monoanhydride.
The acid, referred to as ‘tetra acid’, is prepared as follows: In a Friedel–Crafts reaction, acenaphthene 53 is reacted with malonic dinitrile and aluminium chloride. The resulting condensation product 56 is oxidized with sodium chlorate/hydrochloric acid to form the dichloroacenaphthindandione 57. Oxidation with sodium hypochlorite solution/sodium permanganate affords naphthalene tetracarboxylic acid 49, which mostly exists as the monoanhydride 49a. The dianhydride 49b, on the other hand, evolves only after drying at approx. 150 °C.

A different synthetic route involves halogenation (bromination, chlorination) of pyrene (58), which is thus converted into the tetrahalogen derivative. Oxidation with sulfuric acid to form a diperinaphthindandione with subsequent oxidation, once again in a sodium hydroxide solution [118], yields the tetra sodium salt of naphthalene tetracarboxylic acid 59:

3.4.2.2 Chemistry, Manufacture and Crystal Structures
The synthetic route to perinone pigments, as to perylene pigments, starts from an anhydride, in this case from the monoanhydride of naphthalene tetracarboxylic acid.
The perinone ring system is thus formed by condensation with aromatic o-diamines. Reaction of o-phenylenediamine with the monoanhydride is typically achieved in glacial acetic acid at 120 °C. A mixture of the cis- and trans-isomers evolves, which precipitates as a solid solution (mixed crystal):
Scheme 3.2 Molecular structures of P.R.194 and P.O.43.
By variation of the synthetic conditions, it is possible to enhance the yield of the economically interesting trans-isomer to about 60% [119].
The isomers are separated by taking advantage of the solubility characteristics of their respective salts. By heating the isomer mixture in ethanol/potassium hydroxide, the cis-isomer is dissolved, whereas the trans-isomer is precipitated as a sparingly soluble potassium salt. Satisfactory results are also achieved through fractionation with concentrated sulfuric acid. Either one of these techniques is followed by traditional methods of converting the product into a commercially useful pigment. The options include milling, acid treatment and solvent treatment at elevated temperature.
Other synthetic routes, which exclusively produce the desired trans-isomer, are possible, but have not been implemented industrially, for example [120]:

Figure 3.40 shows the crystal structure of the trans-isomer (P.O.43) [121, 122]. The molecule is situated on a crystallographic inversion centre. Molecules of neighbouring stacks form an interplanar angle of almost 90° (Figures 3.41 and 3.42).
The cis-isomer crystallizes with the same molecular packing as the trans-isomer [122, 123]. The molecule is situated on a crystallographic inversion centre. The molecule itself does not have inversion symmetry; it is disordered on two possible molecular orientations, each with a probability of 50% (Figures 3.43 and 3.44). The atoms of the naphthalene fragment can be superimposed in both orientations, but the positions of the N and O atoms and the positions of the atoms of the terminal phenylene moieties vary according to the molecular orientation. Lattice-energy calculations with dispersion-corrected density functional theory (DFT-D) methods show that the orientation of a given molecule is not fully random, but depends to some extent on the orientation of the neighbouring molecules [124].

Figure 3.40 Crystal structure of P.O.43 [121]: view in the stacking direction.

Figure 3.41 Crystal structure of P.O.43: view perpendicular to the stacks.

Figure 3.42 Crystal structure of P.O.43: one molecule with its neighbours.

Figure 3.43 Disorder of P.R.194 in the crystal [123]. The two possible molecular orientations are drawn in dark and white colours, respectively. The central part of the molecule overlaps for both orientations.
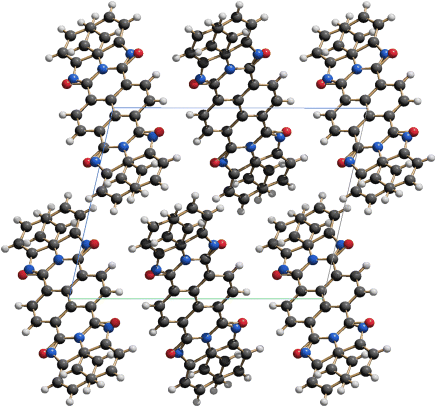
Figure 3.44 Disordered crystal structure of P.R.194, showing all possible atomic positions.
The crystal structure of the solution containing cis- and trans-molecules was investigated by Erich Paulus by single-crystal structure analysis in 1996. However, the data did not allow to locate all atoms [124a]. The same crystal was remeasured 19 years later by Dieter Schollmeyer (Mainz) and Winfried Heyse (Frankfurt), using a more powerful diffractometer, which allowed to determine the crystal structure accurately [124]. In the solid solution, the molecules form the same molecular packing as in the pure cis- and trans-compounds. Each molecular position is either occupied by a trans-molecule, or by a cis-molecule. The cis-molecules are present in two orientations, the trans-molecules in only one, see Figure 3.45. Extensive lattice energy minimisations with DFT-D methods reveal that the distribution of cis- and trans-molecules and their mutual orientations are not fully random, but depend on their neighbours: In stacking direction, a trans-molecule is usually surrounded by two trans-molecules, whereas a cis-molecule prefers to be surrounded by two cis-molecules in the same orientation. The ordering length is calculated to be as large as 230 molecules for a trans-molecule, and 75 molecules for the cis-molecule. In the other spatial directions, cis- and trans-molecules tend to alternate, but the correlation is restricted to a length of 4 to 10 molecules [124].

Figure 3.45 Solid solution of cis- and trans-perinone molecules. The crystal contains cis-molecules in two orientations and trans-molecules in one orientation. In the crystal structure, the atomic positions drawn with open circles have an occupancy of 0.2 only.
The solid solution containing cis- and trans-molecules adopts the same molecular packing as the pure cis- and trans-compounds. Each molecular position in this crystal is either occupied by a trans-molecule, or by a cis-molecule (in either orientation). The distribution of molecules and their mutual orientations are not fully random, but depend on the type and orientation of the neighbouring molecules [124].
The colour of the orange trans-isomer and the red cis-isomer is not only caused by the molecular absorptions bands, but also by exciton interactions, as shown by quantum-mechanical calculations [125]. In a diluted DMSO solution, both compounds are much more yellowish.
3.4.2.3 Properties
Perinone pigments perform very much like perylene pigments. They exhibit shades in the range from orange to bordeaux. Only two members of this group, however, have gained commercial importance. Perinone pigments demonstrate high heat stability and are very lightfast and weatherfast.
3.4.2.4 Commercially Available Perinone Pigments and Their Application
At present, only the two isomers, which constitute the original cis/trans mixture (Vat Red 14), and, to a limited extent, also the mixture itself are commercially used as pigments (Table 3.5).
Table 3.5 Commercially available perinone pigments.
| C.I. Name | C.I. Constitution Number | Structure (see Scheme 3.2) | Shade |
| P.O.43 | 71105 | trans-isomer | reddish orange |
| P.R.194 | 71100 | cis-isomer | bluish red |
| Vat Red 14 | Solid solution of cis- and trans-isomers | scarlet |
3.4.2.4.1 Pigment Orange 43
P.O.43, the trans-isomer, affords a clean reddish shade of orange. Of the two isomers, this is the more important one in its role as a pigment. High cost limits its use to areas that involve high fastness requirements. This is especially true as a colourant for spin dyeing and plastic products, which is the main area of application for P.O.43. Its high lightfastness and weatherfastness is an asset especially in polyacrylonitrile spin dyeing. The pigments are used to colour awnings, tents, canvasses and other items. Samples formulated at 0.3–3% pigment are rated step 7–8 on the Blue Scale for lightfastness, while 0.1% specimen match step 7. The pigment meets high standards in terms of other important fastness properties, such as resistance to dry and wet crocking as well as fastness to chlorohydrocarbons. In the spin dyeing of polyester, P.O.43 withstands the harsh conditions of the condensation process (Section 1.8.3.8). The pigment is also used for the spin dyeing of cellulose acetate and polypropylene fibres and threads. It also lends itself to textile printing.
P.O.43 exhibits good fastness to bleeding in plasticized PVC. Notably, however, bleeding may be observed at extremely low pigment concentrations and at a high plasticizer content. Although P.O.43 is excellently fast to organic solvents and plasticizers, high processing temperatures and low pigment concentrations may lead to blooming (Section 1.6.3.1). In plasticized PVC, like in other media, P.O.43 offers average tinctorial strength. Approximately 0.9% pigment is needed, for instance, to afford 1/3 SD samples containing 5% TiO2. P.O.43 demonstrates excellent lightfastness, even more reduced shades equal step 8 on the Blue Scale. Deep shades, however, darken to a certain extent. No such darkening is observed in corresponding transparent colourations. P.O.43 is also very durable, although it does not satisfy the requirements for long-term exposure.
Transparent and reduced 1/3 SD polyolefin systems are stable up to 300–320 °C. Some 0.25% pigment is required to formulate 1/3 SD HDPE samples containing 1% TiO2. P.O.43, incorporated in injection-moulded partially crystalline plastics such as HDPE, affects the shrinkage of the medium, which precludes its use in non-rotation symmetrical injection-moulded articles (Section 1.6.4.3).
The lightfastness of transparent systems matches step 7–8 on the Blue Scale, depending on the type of plastic and on the pigment content. In white reductions, at 1/3 to 1/100 SD, the pigment scores between step 8 and step 6.
P.O.43 is useful for transparent colourations of polystyrene. At low concentrations (below 0.1%), the pigment dissolves in thermoplastic polyester to produce yellow shades. Even in its dissolved state, the pigment is very lightfast. P.O.43 is inert to peroxides that are typically used to polymerize cast resins based on methacrylic acid methyl esters or unsaturated polyesters. The pigment does not affect the hardening of these polymers.
The paint industry uses P.O.43 primarily as a shading pigment or in white reductions, together with large amounts of TiO2. The pigment is also frequently combined with Molybdate Red pigments. Full shades and similarly deep shades, such as 1 : 10 reductions with TiO2, darken by exposure to light. Considerably high concentrations of TiO2, however, still show excellent lightfastness. Although they do darken to a certain extent upon exposure to weather, this effect is not so noticeable as to affect the applicability of the pigment.
P.O.43 is used especially in industrial paints, including automotive OEM and automotive refinishes, although its significance is somewhat declining. The pigment is used to create metallic effects in paints, especially shades of copper, which are produced by combination with aluminium. There is a certain disadvantage to the fact that the pigment darkens by weathering, even in the presence of UV absorbants. Its heat stability satisfies all types of requirements throughout the paint industry. The pigment is very fast to overcoating and fast to acid, alkali and lime. P.O.43 is also found in emulsion paints. Its excellent weatherfastness makes it a suitable colourant for exterior house paints based on dispersions of synthetic resins. It is mostly used in very strong white reductions.
The printing inks industry uses P.O.43 to colour various types of special-purpose printing inks. The pigment exhibits good heat stability (220 °C for 30 min). Prints containing P.O.43 are fast to sterilization and to a large number of organic solvents and chemicals, as well as to packaged goods. 1/3 SD letterpress proof prints equal step 6 on the Blue Scale for lightfastness, although the shade becomes slightly dull by exposure to light.
In addition, P.O.43 is found in various other media. In wood stains, P.O.43 is frequently used in combination with yellow pigments such as P.Y.83 and carbon black to produce shades of brown. It is also found in leather seasoning systems.
3.4.2.4.2 Pigment Red 194
P.R.194, which is the corresponding cis-isomer, has much less of a commercial impact than the trans-isomer. Its shade is a very dull, bluish red. The pigment is mainly used in paints, especially in architectural paints. It is very lightfast and weatherfast. In contrast to the trans-isomer, P.R.194 does not darken by exposure to light and weather, not even in the full shade range. Although its stability to organic solvents is high, the pigment is not entirely fast to overcoating. This limits its use in certain areas, such as in automotive finishes. P.R.194 is also found in emulsion paints, especially since its excellent weatherfastness makes it a useful product for exterior application. The pigment is fast to acid, alkali and lime.
P.R.194 is also used in textile printing, for instance in combination with carbon black, to produce shades of brown. It is both very lightfast and durable in these media. The colouration equal step 7 on the 8 step weatherfastness scale [126].
P.R.194 is less important in plastics. It shows some bleeding in plasticized PVC. Moreover, the pigment frequently dissolves in its medium as the temperature is increased, even in rigid PVC. It thus exhibits a temperature-dependent colour shift. P.R.194 affords an orange shade as it dissolves in polystyrene, a fact that may present problems in matching defined colours within a certain concentration/temperature range. In polyolefins, the pigment is heat stable up to 270 °C. In some countries, P.R.194 is also an important colourant for polypropylene spin dyeing. Dispersed in polyester during spin dyeing, P.R.194 initially dissolves but recrystallizes as the fibre is thermoset, a process that is associated with an appreciable colour change.
3.4.2.4.3 Vat Red 14
The mixture of P.O.43 and P.R.194, which is a mixed phase consisting of both isomers, is also used as a pigment. Its main market is in spin dyeing, especially of polypropylene. Almost as fast as P.O.43, its colour is referred to as scarlet. The product is thermally stable up to 300 °C.
3.4.3 Semiperinone Pigments and Similar Pigments
Semiperinone pigments are condensation products of naphthalene-1,8-dicarboxylic acid with 1,2-diaminobenzenes. They are synthesized by condensation of naphthalic anhydride with 5,6-diaminobenzimidazolone in a high-boiling solvent [127]:
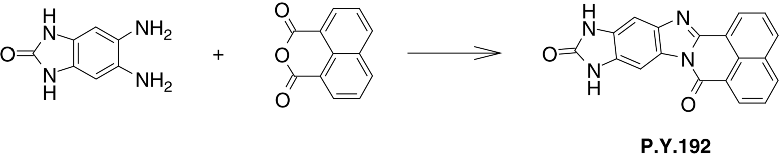
Scheme 3.3 Synthesis of P.Y.192.
Pigment Yellow 192 is listed in the Colour Index under Constitution Number 507300.
P.Y.196 is not a semiperinone pigment, but the condensation product of phthalic acid anhydride with 1,8-diaminonaphthalene, having additionally a phenylsulfonamide group:

Scheme 3.4 Molecular structure of P.Y.196.
P.Y.192 and P.Y.196 are insoluble in water. They are mainly used in polymeric materials such as poly(ethylene terephthalate) (PET), in which the compounds are soluble. This behaviour is more typical for dispersion dyes than for pigments, thus the compounds should be regarded as ‘colourants’ rather than ‘pigments’. Nevertheless they are listed as pigments in the Colour Index.
The main application of P.Y.192 and P.Y.196 is the colouration of polyethylene terephthalate, for PET fibres as well as for PET bottles. The pigments dissolve in the PET, but the high glass transition temperature of PET suppresses the migration. According to the manufacturer, no colouristic changes are observed if P.Y.192 is incorporated in polyester. The pigment maintains its colouristic properties even during the condensation process (Section 1.8.3).
Furthermore, P.Y.192 is a speciality product for the spin dyeing of polyamide. It provides somewhat dull reddish yellow shades. 1/3 SD colourations with and without TiO2 are thermally stable up to 300 °C. Even samples that are formulated at very low pigment concentrations completely maintain their colour. The pigment offers excellent textile fastnesses, such as resistance to dry cleaning and dry heat at 200 °C (Section 1.6.2.4). P.Y.192 is also used in other polymers that are processed at high temperature.
P.Y.196 is an orange-yellow powder produced by high temperature calcination. This pigment has excellent UV and good–fair visible opacity, is chemically inert, heat-resistant and stable to ultraviolet light. It is non-bleeding and non-migratory. It has exceptional durability and hiding power, and is generally used in applications where resistance to heat, light and weather are needed. It is compatible with most resin systems and polymers, and is non-warping.
3.5 Diketopyrrolopyrrole (DPP) Pigments
3.5.1 Chemistry, Manufacture and Crystal Structures
The first synthesis of a diketopyrrolopyrrole pigment was performed unintendedly. In 1974, Farnum et al. attempted to synthesize a 2-azetinone by reacting benzonitrile with bromoacetic acid in the presence of zinc dust. However, “the proposed reaction failed to produce any azetinones”. Instead, in a small yield “a higly insoluble, brilliant red, crystalline compound, C18H12O2N2, mp > 350 °, was isolated from the insoluble residue” [128]. This compound turned out to be 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole, 60:

In the early 1980s, Ciba-Geigy discovered that these compounds could be used as organic pigments, since the brilliant red crystals of 60 turned out to be extremely insoluble. Ciba-Geigy followed up these studies and systematically developed into a synthetic route to a group of very fast red pigments, known as diketopyrrolopyrrole pigments (DPP pigments) [129–131].
DPP pigments are synthesized by reacting succinic ester with benzonitriles in the presence of alcoholate in the corresponding alcohol for base catalysis. Originally starting from sodium methylate/methanol, an important step towards a significantly improved yield was achieved by reaction of succinic tert-alkyl ester in sodium tert-alkyl-alcoholate/tert-alkyl-alcohol.

The intermediates formed during the reaction are not isolated; the synthesis proceeds in one step and affords a very good yield.
Depending on the substituent R4 (CH3, CF3, Cl, Br, N(CH3)2), the resulting reddish yellow to bluish violet pigments show excellent lightfastness and weatherfastness and very good migration fastness. If two differently substituted benzonitriles are used, the product is a mixture of the two symmetrically substituted and of the unsymmetrically substituted diphenyl-DDP pigments (mixed synthesis).
It is possible to manufacture either the transparent or the opaque pigment grade by controlling the particle size during or after synthesis. Small-grained pigment, for instance, can be prepared by adding small amounts of m-phthalonitrile. Other methods of affecting the particle size involve thermal aftertreatment after hydrolysis with optional variation of solvent and pH.
For a given pigment the conversion of the crystal structure into a different crystal modification can lead to a new pigment with a more advantageous shade and improved application properties. Polymorphism has been discovered with some DPP pigments, for example, the β-modification of C.I. Pigment Red 254 [132]. In paint colouration this β-form is significantly more yellowish than the α-form [133].
Crystal structures have been determined for most commercial DPP pigments. In the solid state, the DPP molecule is connected to two neighbouring molecules by two double CO![]() HN hydrogen bonds, leading to chains (Figure 3.46). These hydrogen bonds, together with an efficient molecular packing, result in a good lattice energy and a low solubility, which are responsible for the excellent solvent and migration resistance of this rather small molecule [132]. As soon as the hydrogen bond system is disturbed, for example, by adding a substituent at the nitrogen atom, the compounds become soluble. The red colour of the DPP pigments is apparently a solid-state effect; in solution DPP compounds tend to have a more yellow shade. P.R.254 (R4 = Cl) shows a pale brownish-orange shade in DMSO, with a bright greenish yellow fluorescence under UV light.
HN hydrogen bonds, leading to chains (Figure 3.46). These hydrogen bonds, together with an efficient molecular packing, result in a good lattice energy and a low solubility, which are responsible for the excellent solvent and migration resistance of this rather small molecule [132]. As soon as the hydrogen bond system is disturbed, for example, by adding a substituent at the nitrogen atom, the compounds become soluble. The red colour of the DPP pigments is apparently a solid-state effect; in solution DPP compounds tend to have a more yellow shade. P.R.254 (R4 = Cl) shows a pale brownish-orange shade in DMSO, with a bright greenish yellow fluorescence under UV light.

Figure 3.46 P.R.255: molecular chains formed by CO⋯HN hydrogen bonds.
The DPP molecule is not completely planar. The phenyl rings can be slightly rotated out of the plane. For example, the dihedral angle between the phenyl rings and the central diketopyrrolopyrrole fragment is 7° for P.R.255 (R4 = H), 3° for P.R.254 (R4 = Cl) and 16° for P.R.264 (R4 = Ph).
The arrangement of the molecular chains differs in the individual DPP structures. In the parent compound, P.R.255, the chains are parallel, as in 2,9-dimethylquinacridone (P.R.122) and in the αI-phase of unsubstituted quinacridone (P.V.19) (see Figure 3.47) [134]. In contrast, the chains form a herringbone pattern in P.R.254 (Figure 3.48) [135].

Figure 3.47 P.R.255: arrangement of the chains.

Figure 3.48 P.R.254: herringbone arrangement of chains.
P.R.264 (R4 = Ph) has an unusual helical molecular conformation (Figure 3.49) [136]. The central pyrrolopyrrole moiety exhibits a dihedral angle of 16.2° with the adjacent phenyl rings, which form a dihedral angle of 33.2° with the terminal phenyl rings. The total torsion angle between the terminal phenyl rings is 99.5°. This helical conformation allows the terminal phenyl rings to adopt a herringbone packing (which is preferred for benzene rings), although the central diketopyrrolopyrrole fragments form chains (Figure 3.50). The chains arrange in a parallel packing, as in P.R.255 (Figure 3.51).

Figure 3.49 Helical geometry of P.R.264 in the solid state. The planes of the phenyl rings and of the central diketopyrrolopyrrole (DPP) unit are indicated.

Figure 3.50 P.R.264: Herringbone arrangement of the terminal phenyl rings (drawn in bold) and parallel packing of the central DPP unit (in red).

Figure 3.51 P.R.264, view along the chains.
P.O.73 (R4 = tBu) and P.R.255 (R4 = H) form a mixed crystal with a stoichiometry of 1 : 1. In this rare instance the mixed crystal is ordered, that is, the distribution of molecules is not random (as it would be in a solid solution), but the lattice contains alternatingly one molecule of P.O.73 and one molecule of P.R.255 in a unit cell of doubled size (Figure 3.52). The mixed crystal has the same structure as the non-symmetrical mono-tert-butyl compound (Figure 3.53) [137, 138].
In pure P.O.73 the chains are shifted sidewards to provide space for the bulky tert-butyl groups (Figure 3.54) [139].

Figure 3.52 Mixed crystal of P.O.73 and P.R.255. The mixed crystal is ordered.

Figure 3.53 Crystal structure of the mono-tert-butyl substituted DPP.

Figure 3.54 Crystal structure of P.O.73.
P.O.71 (R3 = CN) exhibits the same molecular packing as the parent compound P.R.255. The additional cyano groups do not disturb the formation of the hydrogen bonds. Cyano groups of neighbouring molecules are arranged antiparallel to optimize the dipole–dipole interactions (Figure 3.55) [140].

Figure 3.55 Crystal structure of P.O.71.
Another path affording novel pigments is a mixed synthesis yielding solid solutions, that is, a mixed crystal with a statistical distribution of different molecules on the positions in the lattice. The mixed synthesis of two DPP pigments can result in ternary solid solutions, which consist of the two symmetrically and the unsymmetrically substituted DPP pigments. Solid solutions have also been described by a combination of DPP and quinacridone pigments. However, the hydrogen bond geometries of DPP and quinacridone molecules are different, and it is questionable, to which extent such solid solutions actually exist.
3.5.2 Properties and Application
Owing to their cleanness of shades often displayed in the range of medium orange to bluish red and their excellent light- and weatherfastness combined with good-to-very good solvent fastness, DPP pigments are primarily used in high-performance applications, for example, in the area of automotive and other industrial paints, for plastics and for printing inks that require outstanding application properties.
3.5.3 Commercially Available DPP Pigments
3.5.3.1 General
Commercially available DPP pigments are summarized in Table 3.6. Within the range of DPP pigments presently offered to the market, P.R.254 plays the most important role. The pigments differ in their substituents and position at the phenyl ring. The p-position is the preferred one; at present only one pigment with a substituent in the m-position is commercially available.
Table 3.6 Commercially available DPP pigments.

|
||||
| C.I. Name | Constitution Number | R3 | R4 | Shade |
| P.O.71 | 561200 | CN | H | medium orange |
| P.O.73 | 561170 | H | C(CH3)3 | medium orange |
| P.O.81 | — | — | — | reddish orange |
| P.R.254 | 56110 | H | Cl | medium red |
| P.R.255 | 561050 | H | H | yellowish red |
| P.R.264 | 561300 | H | C6H5 | bluish red |
| P.R.270 | — | — | — | yellowish red |
| P.R.272 | 561150 | H | CH3 | medium red |
| P.R.283 | — | — | — | yellowish red |
| DPP/quinacridone solid solutions | — | — | — | yellowish to bluish red |
Dependent on the substituents the shade from orange to bluish red follows the order:
DPP/quinacridone and DPP/DPP mixed pigments (‘solid solutions’) are introduced or presently being offered to the market.
3.5.3.2 Pigment Orange 71
Some years ago the DPP pigment was introduced to the market. It produces medium orange shades and is recommended mainly for the use in plastics and printing inks. Special grades for these applications are on the market.
The plastics type is suitable amongst others for HDPE, PP and for PVC. 1/3 SD colourations of HDPE (1% TiO2) require 0.35% pigment. These colourations are heat resistant up to 300 °C and equal step 8 on the Blue Scale for the lightfastness. The shrinkage of the polymer is only slightly affected at temperatures between 220 and 260 °C and somewhat affected at temperatures up to 300 °C. P.O.71 has a very good bleeding fastness in plasticized PVC. It is also of interest for the use in PP fibres.
The special grade for printing inks is recommended for all kinds of printing processes. The prints are very transparent and fast or almost fast to many solvents commonly used in printing inks. The lightfastness is also good.
3.5.3.3 Pigment Orange 73
P.O.73 affords clean shades of medium orange and is suitable to colour baking enamels based on organic solvent systems, powder coatings and coil coating. It is mainly recommended for high grade industrial finishes, especially for automotive finishes. The commercially available type produces opaque coatings, it is suitable for the replacement of lead-free pigment formulations. P.O.73 is less stable to some organic solvents, such as ethanol, methyl ethyl ketone and ethyl acetate, than P.R.254 or P.R.264. Therefore overcoating fastness is not perfect in certain systems.
The heat stability of P.O.73 is good, but deteriorates somewhat in white reductions. The pigment exhibits good weatherfastness, but performs poorer in this respect than P.R.254 and P.R.264.
3.5.3.4 Pigment Orange 81
P.O.81 is a DPP pigment, released some time ago to the market, whose exact chemical constitution has not been disclosed. It affords a very clean orange shade and shows good hiding power, high saturation to achieve deep shade areas, very good weatherfastness and excellent flow and gloss. The pigment can be used in automotive and industrial paints, plastics and industrial and powder coatings.
3.5.3.5 Pigment Red 254
P.R.254, which was introduced into the market as the first representative of DPP pigments, shows good colouristic and fastness properties and has within a short period of time developed into a widely used pigment for high industrial paints, especially in original automotive finishes and automotive refinishes. The commercially available type affords medium shades of red in full shades, while white reductions are somewhat bluish red. The pigment shows good hiding power, it is used primarily in automotive finishes wherever lead-free formulations are required. For economic reasons, P.R.254 is frequently used in combination with the somewhat bluer but less weatherfast opaque type of P.R.170. Combination, for instance, with quinacridone pigments affords opaque shades of bluish red.
P.R.254 shows very good fastness to organic solvents. It is therefore fast to blooming and bleeding in baking enamels. Full shades and similar deep shades are lightfast enough to reach step 8 on the Blue Scale. The pigment also shows very good weatherfastness – a reason for its primary use in original automotive finishes. Its fastness to flocculation can be improved by employing suitable additives.
P.R.254 is also used to colour plastics that are processed at high temperature. A special type has recently been introduced to the market that is used for this purpose. 1/3 SD HDPE colourations of this type are stable up to 300 °C for 5 min. The colourations exhibit high tinctorial strength; their shade is considerably yellower and cleaner than that of the type which is used in paints. 1/3 SD colourations (1% TiO2) require 0.16% pigment. The pigment only affects the shrinkage of the plastic to a minor extent. The lightfastness of 1/3 SD colourations (1% TiO2 and transparent) and of full shades equals step 7–8 on the Blue Scale.
In plasticized PVC, P.R.254 reaches step 8 on the Blue Scale for lightfastness. It shows high tinctorial strength and bleeding fastness.
3.5.3.6 Pigment Red 255
The pigment affords reddish shades of orange, actually scarlet shades, which are distinctly more yellowish than those of P.R.254. The commercially available grade of P.R.255 shows excellent hiding power due to its specific surface area of 15 m2 g−1. Lightfastness and weatherfastness of full shades are excellent, those of tints are very good-to-good, but not quite as good as those of P.R.254. Similar results are obtained for other fastness properties of the two DPP pigments. So, for instance, the stability of P.R.255 to some organic solvents, such as methyl ethyl ketone and ethyl acetate, commonly used in paint systems and finishes, is somewhat inferior compared with P.R.254. Baking enamels are fast to overcoating at common baking temperatures (140 °C, 30 min). Up to 200 °C heat resistance is very good, which is important for the use in powder coatings. Higher temperatures will cause a darkening of the shade.
P.R.255 is recommended for high grade lead-free industrial finishes, especially automotive OEM finishes. The commercially available grade tends to flocculate in many formulations. Additives to improve the fastness to flocculation are available on the market. The rheological behaviour of finishes is characterized by a distinct thixotropy. This can cause problems at the production of mill pastes. P.R.255 is also of interest for the use in tinting systems for decorative paints.
Within the plastics area it can be used in rigid and plasticized PVC. The list of applications includes packaging and decorative printing inks.
3.5.3.7 Pigment Red 264
P.R.264 provides bluish red shades, which are similar to those of P.R.177, an aminoanthraquinone pigment. The fastness properties are similar to the other types of the DPP pigment class. The commercially available grade is distinguished by a very high tinctorial strength. P.R.264 is broad in scope, its main application area is in paints. It is recommended for high grade industrial paints including automotive finishes of different kinds. In metallic and effect finishes it is suitable for very clean red shades. The weatherfastness of the coating is excellent. The coatings are completely fast to overcoating. The flow properties of the commercially available grade are moderate; the pigment exhibits high viscosity in high and medium solids. Special additives increase the tinctorial strength.
The heat stability is very good, which is a prerequisite for the use of P.R.264 in powder coatings and in coil coating. In the plastics industry P.R.264 can be used to colour PVC, and because of its good heat stability also PP, ABS, PA and PET. The tinctorial strength in plastics is very high. The pigment concentration required for 1/3 SD HDPE colourations (1% TiO2) is only 0.09%. The colourations are heat stable up to 300 °C and the corresponding lightfastness equals step 8 on the Blue Scale. The shrinkage of such partially crystalline polymers is only slightly affected. In suitable polymers the weatherfastness is said to be good. The pigment is also recommended for PP dyeing.
In the printing ink area P.R.264 can be used for high grade packaging printing inks. The prints are transparent and show good lightfastness.
3.5.3.8 Pigment Red 270
The pigment was later introduced to the market. The chemical structure has not yet been disclosed.
P.R.270 exhibits a yellowish red shade and is recommended for the colouration of plastics and printing inks.
3.5.3.9 Pigment Red 272
This pigment has been published in the Colour Index. According to the data released by the producer, it is a medium red shade pigment and exhibits high colour strength. P.R.272 is said to be designed for use in PE, PP and PVC plastics applications, such as injection moulding, sheet and cable extrusion and calendering as well as PP fibres application. The heat stability in these plastic materials reaches 300 °C in the mass tone and 260 °C in white reductions with a very low influence on shrinkage in HDPE. The pigment demonstrates good lightfastness and weatherfastness, but mass tone colourations tend to darken.
Owing to its good opacity, the pigment is recommended in general industrial paint applications for lead- and cadmium-free formulations.
3.5.3.10 Pigment Red 283
P.R.283 is a DPP Pigment of which the chemical structure has not yet been disclosed. It presents a strong yellowish red shade, outstanding thermostability and very high transparency and is preferably applicable for the colouration of plastics. The pigment shows no nucleation in plastics and therefore no distortion in moulded plastics, such as HDPE.
3.5.3.11 DPP/Quinacridone Mixed Crystal Phase (‘Solid Solutions’) Pigments
Some of these pigment types which are claimed to be solid solutions, have been released to the market. The commercially available grades afford red shades, which are much bluer than the DPP pigments and much yellower than the quinacridone pigments. The ratio of the two components within the pigment crystal and the physical properties, such as particle size distribution and crystallinity, determine the colouristic and fastness properties. The commercially available grades are recommended for high grade industrial finishes, especially automotive finishes. They are transparent, which is a prerequisite for metallic and effect automotive OEM finishes. In such systems they produce very brilliant red shades with special flop effects (see Section 3.1.5.1.2.4). Pigmented water-reducible base coat systems form stable dispersions. The fastness of these mixed crystal phase pigments to some organic solvents is not perfect. So, for instance, they are somewhat soluble in butyl-glycol; the resulting solutions are fluorescing more or less. Connected with this behaviour is a decrease of the overcoating fastness of the two basic pigments, the DPP pigment and the quinacridone pigment.
The plastics industry uses these pigments mainly for polyolefins. The tinctorial strength is comparatively moderate. 1/3 SD HDPE colourations (1% TiO2), for instance, require between 0.22% and 0.7% of these pigment grades. Such colourations are heat resistant up to 300 °C. Transparent colourations in 1/3 SD are stable up to 250 °C.
3.6 Indigo, Thioindigo and Thiazine Indigo Pigments
Indigo is the oldest of all vat dyes. During its history as a commercial pigment, it was used especially in rubber. Its structure was first described in 1883 by A.v. Baeyer. Knowledge of this structure, together with the development of an industrial-scale synthesis, facilitated the development of a series of indigo-based colourants:

Scheme 3.5 Molecular structure of Indigo (P.B.66), Thioindigo and Thiazine Indigo.
Thioindigo pigments have been known since 1905.
Thiazine indigo pigments differ from thioindigo pigments by additional NH groups, and were first described in 1971.
3.6.1 Indigo
Indigo has been known as a dye since ancient times. It was produced from plants, especially from indigofera. The first fully chemical synthesis was carried out by Baeyer in 1870 [141]. Today, indigo is synthetically produced in large amounts. Indigo is mainly used as a vat dye, for example, for making blue jeans. To a minor extent, it is used as a pigment (Pigment Blue 66).
The bromo derivative 6,6′-dibromoindigo is the main ingredient of natural purple.
3.6.1.1 Crystal Structure
In indigo, the NH group donates a bifurcated intra-/inter-molecular hydrogen bond to the keto group of the same molecule and to a keto group of a neighbouring molecule (Figure 3.56). Each molecule is connected to four different neighbouring molecules, resulting in a criss-cross pattern as in the γ-phase of P.V.19 (quinacridone).

Figure 3.56 Indigo [142]: hydrogen bond system (a) and criss-cross pattern (b).
3.6.1.2 Properties and Application
Unsubstituted indigo is marketed as Pigment Blue 66, 73000. It is a suitable pigment for spin dyeing of synthetic viscose rayon and spun rayon, to which it lends navy blue shades. The pigment provides good lightfastness: the samples match step 5–6 to step 7 on the Blue Scale, depending on the standard depth of shade. However, Pigment Blue 66 performs poorly regarding several textile fastnesses, especially in terms of bleaching with chlorite, and vat resistance.
3.6.2 Thioindigo Pigments
The parent structure of all thioindigo pigments is the unsubstituted thioindigo molecule (Friedländer, 1905), which is of little technical importance. More important products are chlorinated and/or methylated thioindigo derivatives, which were developed in subsequent years. During the 1950s, several thioindigo derivatives gained commercial recognition as pigments. This was only after it became possible to appropriately finish the crude products after manufacture. Manufacture of most of these pigments has been discontinued.
3.6.2.1 Chemistry, Manufacture and Crystal Structures
Thioindigo pigments have the following general structure:

In commercially important products, R typically represents a chlorine atom or a methyl group, and n stands for 2 or 3. The compounds assume the trans structure.
The synthesis is performed in two steps [143, 144]:
- attaching the five-membered ring to the benzene ring;
- oxidatively linking two molecules of the resulting thionaphthen-3-one.
The most important route involves cyclization of appropriately substituted phenylthioglycolic acids:

Suitable agents are chlorosulfonic acid monohydrate, or concentrated sulfuric acid. Other methods include cyclization of phenylthioglycolic acid chlorides, which is afforded by Friedel–Crafts reaction with aluminium chloride, possibly in the presence of sulfuric acid/sodium chloride. The acid chloride is obtained from phenylthioglycolic acid with thionyl chloride or phosphorus trichloride.
There are more routes to cyclization. Phenylmercaptoacetic acid may be replaced by its o-carboxy or o-amino derivative to form the thionaphthen-3-one. The first reaction is performed in molten alkali, the second proceeds via diazotization, Sandmeyer reaction with sodium cyanide/copper cyanide, alkaline hydrolysis and acid treatment.
Oxidation may be achieved in the presence of oxygen or air. Other suitable oxidants include sulfur, sodium polysulfide, iron(III)chloride, potassium ferrocyanide(III) or potassium dichromate, peroxydisulfate or salts of aromatic nitrosulfonic acids. An aqueous/alkaline medium is used in the presence of a high boiling organic solvent that is not miscible with water or which is almost immiscible with water. Cyclization with chlorosulfonic acid can be followed directly by oxidation with bromine to afford the thioindigo system, without separation of the intermediate.
The synthetic route to tetrachlorothioindigo may serve as an example for an industrial-scale synthesis. Starting material is 2,5-dichlorothiophenol, which is obtained by reduction of 2,5-dichlorobenzene sulfochloride or by reaction of the 2,5-dichlorobenzene diazonium salt with potassium-o-ethyldithiocarbonate and hydrolysis or with disodium sulfide and subsequent reduction:

2,5-Dichlorothiophenol is treated with chloroacetic acid to form 2,5-dichlorophenylmercaptoacetic acid. Cyclization followed by oxidation with chlorosulfonic acid at 35 °C affords 4,7-dichlorothionaphthen-3-one. Direct addition of bromine in chlorosulfonic acid without separating the intermediate yields the 4,4′,7,7′-tetrachlorothioindigo derivative through oxidative dimerization. It is also possible to separate the monoheterocycle and to oxidize it with oxygen in an alkaline medium:

3.6.2.1.1 Aftertreatment
Tetrachlorothioindigo, by far the most important member of this pigment series, will serve as a frame of reference.
Although there is one route [145] that describes the direct synthesis of the tetrachlorothioindigo pigment by oxidation of 3-hydroxy-4,7-dichlorothionaphthenone with oxygen in an aqueous alkaline medium, this is somewhat of an exception. In most cases, it is necessary to modify the crude thioindigo derivative by appropriate aftertreatment to develop the desired pigment properties.
The list of options includes milling the crude product with salt or dispersing agents or reprecipitation from sulfuric acid or chlorosulfonic acid, followed by aftertreatment with organic solvents [146]. Tinctorially strong transparent pigment grades, for instance, are obtained by milling the crude pigment suspension in the presence of an aqueous base [147]. The same result is achieved by oxidizing the leuco form of tetrachlorothioindigo in the presence of sodium dithionite with air while applying shearing forces (for instance in a pearl mill) [148].
3.6.2.1.2 Crystal Structures
Thioindigo pigments cannot form hydrogen bonds in the solid state. Figure 3.57 shows the molecular packing in P.R.88.

Figure 3.57 Crystal structure of P.R.88 [149]. The sulfur atom and the carbonyl groups are disordered with occupancies 0.65 : 0.35. The minor occupied positions of S and CO are shown as open circles.
3.6.2.2 Properties and Application
Thioindigo pigments provide a range of hues from red violet and maroon to brown shades. The type and position of the substituents affects the shade.
Electron-donating substituents (methyl groups) at the 5-position cause the most drastic bathochromic shift, which decreases in the order: 4->7->6-position. The reverse is also true in the case of electron acceptors (such as chlorine): they cause more of a bathochromic shift in the 6-position than in the 5-position.
As a class, tetrachlorothioindigo pigments exhibit good-to-excellent lightfastness and weatherfastness as well as solvent stability and migration fastness. 4,4′,7,7′-Substituted derivatives demonstrate highest fastness to solvents. Chlorine substitution has a better effect in this respect than methyl substitution. Shifting only one of the substituents on 4,4′,7,7′-tetrachlorothioindigo, for instance, will adversely affect the migration resistance of the parent compound (fastness to bleeding).
3.6.2.3 Commercially Available Thioindigo Pigments and Their Application
3.6.2.3.1 General
Of the two remaining thioindigo pigments, only Pigment Red 88, the 4,4′,7,7′-tetrachlorothioindigo, has been able to maintain interest throughout the pigments industry. It is broad in scope. The other type, Pigment Red 181, is a special-purpose type for a limited number of applications. Table 3.7 lists the chemical constitutions of both pigments.
Table 3.7 Commercially available thioindigo pigments.

|
||||||
| C.I. Name | C.I. Constitution Number | R4 | R5 | R6 | R7 | Shade |
| P.V.38 | 73395 | CH3 | Cl | H | CH3 | reddish purple |
| P.R.88 | 73312 | Cl | H | H | Cl | red-violet |
| P.R.181 | 73360 | CH3 | H | Cl | H | bluish red |
Unsubstituted thioindigo, which is marketed as Vat Red 41, 73300, is used in rigid PVC, polystyrene and several other plastics. Dissolved in its medium, the pigment affords fluorescent bluish red shades.
Unsubstituted indigo is marketed as Pigment Blue 66, C.I. 73000. It is a suitable pigment for spin dyeing of synthetic viscose rayon, to which it lends navy blue shades. The pigment provides good lightfastness: the samples match step 5–6 to step 7 on the Blue Scale, depending on the standard depth of shade. However, pigment Blue 66 performs poorly regarding several textile fastnesses, especially in terms of bleaching with chlorite, and vat resistance.
3.6.2.3.2 Pigment Red 88
P.R.88 has stimulated considerable interest throughout the paint industry. It is a suitable colourant especially for industrial coatings, including automotive (OEM) finishes and automotive refinishes. Its red–violet shade is used frequently to produce opaque dark shades of red, bordeaux and maroon, for which purpose the pigment is typically used in combination with inorganic pigments, such as Iron Oxide Red or Molybdate Red. P.R.88 is both very lightfast and weatherfast, although in white reductions and as a shading pigment it fails to reach the standards of P.V.19, the β-modification of unsubstituted quinacridone, which covers the same range of shades. Moreover, β-P.V.19 performs much better in pigment combinations than P.R.88, and it provides much cleaner shades in such combinations.
Deep shades of maroon tend to form water spots in certain binder systems, especially in media that are based on acrylic resin. More or less distinctive light spots appear on the coating. The effects that cause this phenomenon remain to be elucidated. Factors such as long-term weathering at elevated temperature, UV radiation and the presence of demineralized water probably cause reduction and solvation effects within the coating. Products are available that are much less susceptible to these agents. Rub-out effects, especially flocculation, may also present problems in various binder systems. Special-purpose grades are therefore available that are more stable to flocculation.
P.R.88 shows good stability to organic solvents. Its fastness to overcoating is good, but not reliable at baking temperatures above 140 °C. Paints containing P.R.88 are acid and alkali resistant.
P.R.88 is also found in other systems throughout the paint industry, such as in various types of air drying systems and powder coatings.
P.R.88 is one of the standard red–violet pigments for use in plastics. It is particularly suitable for use in PVC, including PVC plastisols and PUR coatings. The pigment shows a more or less pronounced tendency to migrate, depending on the type. New types have been introduced recently that are much faster to bleeding. As a pigment for plasticized PVC, P.R.88 is in direct competition with Pigment Violet 32, which affords a very similar shade, has the advantage of being fast to bleeding and demonstrates considerably higher tinctorial strength. In light tint, however, P.V.32 is somewhat less lightfast than P.R.88. P.R.88 shows excellent lightfastness: transparent and white reductions at 1/1 to 1/25 SD equal step 8 to step 6 on the Blue Scale, depending on the method of stabilization. The pigment is also very weatherfast, but fails to satisfy the requirements for long-term exposure.
Transparent polyolefin systems containing P.R.88 are stable up to 260–300 °C, depending on the type and on the pigment content. 1/3 SD systems are heat stable up to approximately 240–260 °C. Some types are recommended only for use in LDPE at low processing temperatures. The lightfastness of such specimens is between step 6 and step 7 on the Blue Scale. P.R.88 considerably affects the shrinkage of injection-moulded articles, a feature which somewhat restricts its application in such systems.
P.R.88 is recommended for use in PP spin dyeing, special-purpose grades are used for polyacrylonitrile spin dyeing. The resulting fibres are completely fast to perspiration, dry and wet crocking, perchloroethylene, and other important solvents, as well as to dry heat fixation. In the concentration range between 0.1% and 3.0%, the pigment equals step 7 and step 7–8 on the Blue Scale, respectively. P.R.88 meets the performance standards for use in canvasses, tents and similar articles. Used in the spin dyeing of polyester, the pigment dissolves in the fibre. Both sublimation and migration fastness are excellent in this medium. P.R.88 is not always suitable for use in polystyrene. Depending on the temperature, the pigment dissolves in this polymer and accordingly changes its colour and fastness properties. This presents major difficulties in trying to match a defined shade.
Incorporated in methacrylate and unsaturated polyester cast resins, P.R.88 not only withstands several hours of thermal exposure during processing but is also resistant to the peroxides that are used as catalysts. Some types accelerate the polymerization process, that is, the hardening of the plastic.
P.R.88 is also used for printing inks. Its red–violet shade is especially used for printing inks that are to be targeted for packaging, posters and other special purposes. The prints are entirely fast to organic solvents, plasticizers and packaged goods, such as butter and soap. P.R.88 prints are fast to alkali and acid, heat stable up to 200 °C, and fast to sterilization.
The list of applications also includes decorative printing inks for laminated plastic sheets based on melamine or polyester resin. The required fastness and performance properties are very good: the pigment does not, for instance, colour the clear sheets. 8% gravure prints (20 µm cell depth) equal step 8 on the Blue Scale for lightfastness.
P.R.88 is also used in wood stains, for instance in combination with yellow pigments, such as P.Y.83, or carbon black to produce shades of brown.
3.6.2.3.3 Pigment Red 181
P.R.181 is a special-purpose product for polystyrene and similar polymers (Section 1.8.3.3). P.R.181 largely dissolves in its medium at the high temperatures at which these polymers are processed. The pigment affords brilliant, bluish shades of red. It possesses excellent lightfastness and satisfies the application requirements.
Another important market is in toothpaste, for which the pigment has been approved worldwide. It also lends colour to lipstick and other decorative cosmetics. In the USA, it is registered by FDA as D&C Red 30, in Germany as C-Rot 28 according to the DFG Catalogue for Cosmetic Colourants, and in Japan as Red No. 226.
3.6.2.3.4 Pigment Violet 38
Pigment Violet 38 is produced from 2,5-dimethyl-4-chlorophenyl-mercaptoacetic acid by oxidation with chlorosulfonic acid. The pigment has a red light purple shade. It is used in cotton yarn dyeing indigo with bromine, direct printing of cotton fabrics. The pigment resists printing and discharging printing.
3.6.3 Thiazine Indigo Pigments
Thiazine indigo pigments were invented around 1970. In a patent [150] more than 100 derivatives were claimed, including a description of their colours, but in fact only a few of them have been actually synthesized at that time. After work on thiazine indigo compounds has been suspended for about 20 years, development recommenced in the mid-1990s, and terminated several years later. Two compounds were registered in the Colour Index, but no thiazine indigo compound has ever been produced on an industrial scale, because the fastness properties hardly justify the synthetic effort.
3.6.3.1 Chemistry, Manufacture and Crystal Structures
Thiazine indigo pigments (THI pigments) have the general formula:

Thiazine indigo pigments are produced by condensation of o-thioanilines with dihalogenated maleic acid anhydrate. The synthesis yields thiazine-indigo derivatives in their cis-isomeric form, which are thermally transformed into the thermodynamically more stable trans-isomer:

In the solid state, THI compounds form chain structures via double hydrogen bonds [151] (Figure 3.58). Different arrangements of chains are possible, for example, parallel, antiparallel or with a herringbone pattern. Such molecular chains built by double hydrogen bonds are also found in DPP pigments and in several quinacridone phases. The arrangement of chains can be similar, too. For example P.O.80 (unsubstituted THI) has the same herringbone arrangement of molecular chains as P.R.254 (4,4′-dichloro-DPP), whereas P.R.279 (7,7′-dichloro-THI) has the same layered arrangement of chains as P.R.255 (unsubstituted DPP) and P.R.122 (2,9-dimethylquinacridone).

Figure 3.58 Crystal structure of P.R.279 [152]: (a) molecular chains connected by hydrogen bonding; (b) view along the chains. Chlorine atoms are shown in green, sulfur in yellow.
3.6.3.2 Properties
Thiazine indigo pigments offer yellow, orange, red or brown shades, depending on the substitution patterns and on the polymorphic form. For example, substitution with Cl atoms at positions 7 and 7′ results in a red pigment (P.R.279), whereas the compound with CF3 groups at positions 6 and 6′ is yellow. According to preliminary investigations, thiazine indigo pigments have a high refractive index (strongly anisotropic, between η = 1.55 and about 2.1, on average η = 1.81) [151], which explains their outstanding hiding power. The fastness properties of thiazine indigo pigments are not as high as those of for example DPP pigments.
3.6.3.3 Commercially Available Thiazine Indigo Pigments and Their Application
In the Colour Index two thiazine indigo derivatives are registered: P.O.80 and P.R.279 (Table 3.8).
Table 3.8 Commercially available thiazine indigo pigments.

|
|||
| C.I. Name | C.I. Constitution Number | R7,R7′ | Shade |
| P.O.80 | 35714 | H | reddish orange |
| P.R.279 | — | Cl | yellowish red |
3.6.3.3.1 Pigment Orange 80
Pigment Orange 80 is recommended to be used for the colouration of paints and plastics, especially for the mass colouration of polyethylene. It has particularly very high opacity and a very bright colouration in paint applications.
3.6.3.3.2 Pigment Red 279
P.R.279 is a bright yellowish red pigment with outstanding hiding power. Lightfastness and weatherfastness are restricted to the full shade area, in white reductions the pigment shows distinct fading. P.R.279 is resistant to the major solvents used in paint applications. The pigment can be used in industrial paints, where the high opacity and cleanliness of shades makes it very useful in the replacement of lead containing pigments. According to the high heat stability P.R.279 is suitable for powder and coil coating applications, but due to the restricted light and weatherfastness it is mainly used for indoor purposes.
3.7 Various Polycyclic Pigments Derived from Anthraquinone
This section refers to pigments derived from the anthraquinone skeleton, either by chemical structure or by synthesis. The complex fused-ring systems that are discussed in this context are all at least remotely related to the parent anthraquinone structure.
Here, these polycyclic pigments are classified by chemical constitution. The resulting classes include aminoanthraquinones, hydroxyanthraquinones, heterocyclic and polycarbocyclic anthraquinone pigments.
Most listed compounds have known a long history as vat dyes (Section 3.3) before their pigment properties gained commercial recognition.
3.7.1 Aminoanthraquinone Pigments
As a group, these pigments include derivatives of 1-aminoanthraquinone, which is the only derivative of the possible parent compounds to have stimulated commercial interest. While the synthetic routes to the pigment derivatives are found under the respective pigments, the pathway for 1-aminoanthraquinone will be described separately.
3.7.1.1 Synthesis of 1-Aminoanthraquinone
The traditionally most important route to 1-aminoanthraquinone [153] proceeds via nucleophilic exchange of anthraquinone-1-sulfonic acid or 1-chloroanthraquinone with ammonia. Replacing ammonia by amines affords the corresponding alkyl or arylaminoanthraquinones.
Sulfonation of anthraquinone to form the 1-sulfonic acid is achieved at approximately 120 °C with 20% oleum in the presence of mercury or a mercury salt as a catalyst [154]. Without this catalyst, the reaction produces the 2-sulfonic acid. Exchange with aqueous ammonia (30%) at about 175 °C under pressure converts the potassium salt of 1-sulfonic acid into 1-aminoanthraquinone in 70–80% yield. To avoid sulfite formation, the reaction is performed in the presence of an oxidant, such as m-nitrobenzosulfonic acid, which destroys sulfite.
A less important pathway for 1-aminoanthraquinone involves replacing the chlorine atom of 1-chloroanthraquinone by an amino group. The displacement is achieved with an excess amount of aqueous ammonia at about 200–250 °C in the presence of acid-binding agents.
Nitration of anthraquinone with a stoichiometric amount of nitric acid in sulfuric acid affords 1-nitroanthraquinone.
This reaction, which has been known for more than 100 years, has been reinvestigated extensively in an attempt to furnish a purer reaction product and to improve the yield. Interrupting the nitration at approximately 80% completion by removing 1-nitroanthraquinone by distillation makes it possible to separate the side products, which remain in the distillation residue.
The subsequent reaction of the pure 1-nitroanthraquinone to 1-aminoanthraquinone used to be carried out mainly by reaction with sodium sulfides in an alkaline medium and is now performed by nucleophilic replacement of the nitro group with ammonia in organic solvents, affording up to 98% yield. The overall reaction affords approximately 70% yield, in comparison to the roughly 50% yielded by the ‘classical method’ that proceeds via the 1-sulfonic acid. Moreover, the newer method is also superior to the old one because recyclization of the solvent makes it ecologically more attractive.
3.7.1.2 Anthraquinone-Hydrazone Pigments
1-Aminoanthraquinone has only little importance as a diazo component for corresponding hydrazone pigments. Diazotization of 1-aminoanthraquinone with subsequent coupling onto different coupling components, which, especially by heterocyclic groups with additional carbonamide groups, leads to highly insoluble, migration fast pigments, has been described in several patents. This synthesis produces yellow to red monohydrazone pigments, which are recommended primarily for use in plastics.
1-Aminoanthraquinone is diazotized advantageously with nitrosylsulfuric acid after being dissolved in sulfuric acid. Coupling the diazo compound onto barbituric acid, for instance, affords a yellow pigment [155] with the structure 61:

Coupling 1-aminoanthraquinone with certain Naphthol AS derivatives produces red pigments [156] with the structure 62, which are suitable colourants for plastics:

The compounds are obtained by coupling diazotized 1-aminoanthraquinone onto 2-hydroxy-3-naphthoic acid, followed by separation, drying and conversion into the hydrazone dye acid chloride. Condensation with amines (structure 63) is achieved in an aprotic organic solvent:

Coupling 1-aminoanthraquinone onto pyrazolo[5,1-b]quinazolone with the general structure:
![Figure depicting the coupling 1-aminoanthraquinone onto pyrazolo [5,1-b] quinazolone with the general structure.](http://images-20200215.ebookreading.net/4/1/1/9783527326082/9783527326082__industrial-organic-pigments__9783527326082__images__c03__n3cs041.gif)
primarily affords red pigments with very good fastness properties. R represents a methyl or phenyl group and R′ stands for various substituents, but primarily for hydrogen, chlorine or bromine. This point is illustrated by Pigment Red 251, C.I. 12925 [157] as follows:

Scheme 3.6 Molecular structure of P.R.251.
Other anthraquinone-hydrazone pigments are obtained by coupling 1-aminoanthraquinone derivatives onto bis-quinazolinone methanes with the following chemical structure (64):

Compound 64 is prepared through condensation of, possibly substituted, anthranilic acid amides with malonic diethyl esters in the presence of pyridine. Diazotization of 1-amino-5-benzoylaminoanthraquinone, which is then coupled onto 64, for instance, affords an orange pigment with the structure 65 [158]:

3.7.1.3 Other Aminoanthraquinone Pigments
As a class, these pigments include 1-aminoanthraquinone compounds that bear free amino groups, and also derivatives in which the primary amino group is substituted by an aryl or a heteroaryl moiety. Very few representatives are industrially important.
Formation of CC links in the anthraquinone molecule, especially substitutions with aryl moieties, proceed via copper-catalysed nucleophilic exchange of halogenated anthraquinone compounds. These methods include dimerization of 1-aminoanthraquinone.
Pigment Red 177, a red pigment that is listed in the Colour Index under Constitution No. 65300, has the chemical structure of 4,4′-diamino-1,1′-dianthraquinonyl (66) [159]:

Scheme 3.7 Molecular structure of P.R.177.
Simultaneously with Hansa Yellow G, compound 66 was first described as early as 1909 by Farbenfabriken Bayer. Its preparation starts from 1-amino-4-bromoanthraquinone-2-sulfonic acid (bromamine acid) (67). Dimerization is achieved through the Ullmann reaction, that is, treatment with fine-grain copper powder in dilute sulfuric acid at 75 °C. The separated intermediate, the disodium salt of 4,4′-diamino-1,1′-dianthraquinonyl-3,3′-disulfonic acid (68), is heated to 135–140 °C in the presence of 80% sulfuric acid to cleave the sulfonic acid groups [160]:

The result is a bluish red pigment, the only one amongst the commercial products that possesses free amino groups.
X-Ray diffraction analysis of P.R.177 disclosed a twisting of the two anthraquinone units by 75° relative to each other [161, 162]. The amino group forms one intramolecular and one intermolecular hydrogen bond (Figures 3.59 and 3.60).

Figure 3.59 Molecular structure of P.R.177 in the solid state [162].

Figure 3.60 Crystal structure of P.R.177, showing six molecules with their hydrogen bond pattern.
Pigment Yellow 199, Constitution Number 653200, can be considered as a dicarbonamide of P.R.177 with both amino groups substituted by lauric acid (C11H23COOH):

Scheme 3.8 Molecular structure of P.Y.199.
Acylation of 1-aminoanthraquinone affords 1-acylaminoanthraquinones. The carbonamide function renders these compounds very fast to migration. The migration fastness may be improved further by introducing substituents that carry additional carbonamide groups, or by molecular dimerization using aromatic dichlorides.
The most important route to 1-acylaminoanthraquinones involves reaction of 1-aminoanthraquinone with acid chlorides in an organic solvent. Reaction of 1-aminoanthraquinone with benzoyl chloride in nitrobenzene at 100–150 °C affords 1-benzoylaminoanthraquinone, a yellow pigment that is registered as Colour Index Constitution No. 60515, but has no C.I. name. The reaction may also be performed in the presence of a tertiary amine, which acts as a proton acceptor:

Benzoylation of 1-amino-4-hydroxyanthraquinone affords a bluish red pigment, which is known as Pigment Red 89, C.I. 60745:

Scheme 3.9 Molecular structure of P.R.89.
1-Amino-4-hydroxyanthraquinone may be obtained from 1-nitro-4-hydroxyanthraquinone by reduction with sodium sulfide.
Condensation of 1-aminoanthraquinone with phthalic acid chloride in o-dichlorobenzene at 145 °C also yields a yellow pigment, registered as Pigment Yellow 123, 65049 [163]:

Scheme 3.10 Synthesis of P.Y.123.
Its meta-isomer is Pigment Yellow 202, Constitution number 65410:

Scheme 3.11 Molecular structure of P.Y.202.
It exhibits a dull greenish yellow shade and is recommended to be used for the colouration of PVC.
The para-isomer, Pigment Yellow 193, C.I. 65412, provides a reddish yellow shade:

Scheme 3.12 Molecular structure of P.Y.193.
The four pigments P.R.89, P.Y.123, P.Y.202 and P.Y.193, however, currently lack significant commercial use.
Amongst heterocyclic substituted 1-aminoanthraquinone derivatives, the 2 : 1 reaction product with 1-phenyl-2,4,6-triazine, also referred to as Pigment Yellow 147, 60645, should be mentioned as an example [164]. It is a reddish yellow pigment with the chemical structure 69:

Scheme 3.13 Molecular structure of P.Y.147.
This pigment is prepared by condensation of two equivalents of 1-aminoanthraquinone with 2,4-dichloro-6-phenyl-1,3,5-triazine in the presence of a base in an organic medium. It is also possible and even preferable to treat one equivalent of phenylguanamine with two equivalents of 1-halogenanthraquinone in the presence of an additional tertiary base with copper(I)iodide as a catalyst. In contrast to copper iodide itself, the corresponding addition compounds have the advantage of dissolving easily in organic solvents, which facilitates their separation:

Phenylguanamine (2,4-diamino-6-phenyl-1,3,5-triazine) is synthesized from benzonitrile and dicyanamide:

3.7.1.4 Commercially Available Aminoanthraquinone Pigments
3.7.1.4.1 Pigment Yellow 147
P.Y.147 (Scheme 3.13) provides a medium to reddish shade of yellow. It is a speciality product for use in polystyrene. Colourations up to 1/3 SD are heat stable up to 300 °C. In terms of lightfastness, these samples match step 7 on the Blue Scale. Incorporated in polyolefins, P.Y.147 is also stable to temperatures up to 300 °C. The pigment is comparatively weak: 0.35% P.Y.147 is required to afford 1/3 SD HDPE samples (1% TiO2).
3.7.1.4.2 Pigment Yellow 193
P.Y.193 (formula see Scheme 3.12), an anthraquinone pigment which, so far, is produced in Japan has gained only little importance. According to the producer's information the pigment is utilized in printing inks and in plastics.
P.Y.193 produces dull reddish yellow shades. In printing inks the tinctorial strength is extremely weak. Thus, in offset printing inks it exhibits only about half of the tinctorial strength of the weak P.Y.191 in a distinctly redder shade, and compared with the diarylide pigment P.Y.83 it produces only about a sixth of the strength.
P.Y.193 is tinctorially equal weak in plastics. Colourations of HDPE in 1/3 SD (1% TiO2) require 0.43% pigment; the heat stability of the colourations reaches 300 °C.
The pigment shows good lightfastness. Its tinctorial strength in plasticized PVC P.Y.193 is distinctly weaker than P.Y.191.
3.7.1.4.3 Pigment Yellow 199
The formula of P.Y.199 is shown in Scheme 3.8. The commercially available grade is especially of interest for the use in PP spin dyeing. It is characterized by a very small particle size distribution and, thus, connected with very high gloss of the colourations. P.Y.199 is also recommended for mass dyeing of polyolefins and other polymers.
Some 0.22% pigment is required to afford 1/3 SD samples of HDPE (1% TiO2) and more than 240 °C are necessary to develop that tinctorial strength. The heat stability in polyolefins reaches 300 °C. Owing to only moderate migration properties P.Y.199 is unsuitable for the colouration of plasticized PVC.
3.7.1.4.4 Pigment Red 89
The pigment used to be marketed, but its production has now been discontinued.
P.R.89 (Formula see Scheme 3.9) affords a very bluish shade of red, referred to as pink. It exhibits good tinctorial strength and good lightfastness. Full shades in a combination nitro lacquer, for instance, equal step 7–8 on the Blue Scale. Somewhat poor overall fastness restricts the applicability of P.R.89. The pigment was considered a special-purpose product for artists' colours. It used to be employed also in spin dyeing viscose products.
3.7.1.4.5 Pigment Red 177
P.R.177 (Formula see Scheme 3.7) is mainly applied in industrial paints, in spin dyeing and in polyolefin and PVC colouration.
The paint industry uses P.R.177 primarily in combination with inorganic pigments, especially with Molybdate Red Pigments. The resulting colourations have the advantage of showing high brilliance and cleanness in shades that are not accessible with other organic pigments. Combined with inorganic pigments, P.R.177 exhibits high lightfastness and weatherfastness. Combinations with Molybdate Red are also used in automotive finishes, especially for automotive OEM finishes and for automotive refinishes. The types that have been marketed thus far are highly transparent, which makes them suitable colourants for transparent paints. In metallic effect coatings, P.R.177 is not weatherfast enough to meet higher standards of application. In an improperly formulated paint system, the pigment may react with aluminium or other reducing agents. Its weatherfastness decreases rapidly in white reductions. At typical processing temperatures for oven drying systems, P.R.177 is entirely fast to overcoating.
A very opaque version has been introduced to the market in the USA. This type, which is also yellower than traditional varieties, demonstrates not only excellent rheological behaviour but also high flocculation stability and therefore high gloss. It is slightly more weatherfast than more transparent types. The main application is in lead-free automotive finishes for full shades.
P.R.177 shows excellent heat stability in plastics. 1/3 SD HDPE samples containing 1% TiO2, for instance, are fast to up to 300 °C. Nucleation is not observed, that is the pigment does not affect the shrinkage of injection-moulded polymer articles. In terms of tinctorial strength, P.R.177 is comparatively weak in these media: 0.3% pigment is needed to afford 1/3 SD colourations containing 1% TiO2. P.R.177 systems are less lightfast than those containing other pigments of this class. P.R.177 is not entirely fast to bleeding in plasticized PVC.
Good transparency makes the grades with fine particle sizes suitable colourants for transparent films. The products exhibit excellent lightfastness. P.R.177, in combination with Molybdate Red pigments, provides better properties than other organic red pigments. Suitable media, also for such pigment combinations, are PUR and PVC coatings. In addition, P.R.177 is also used for spin dyeing polypropylene, polyacrylonitrile and polyamide.
The printing ink industry uses P.R.177 primarily to print securities, especially bank notes.
P.R.177 is also used for the filters of liquid crystal displays [165].
3.7.1.4.6 Pigment Red 251
P.R.251 (Formula see Scheme 3.6), a yellowish red pigment, is marketed as a variety with a very coarse particle size, which exhibits good hiding power and is recommended particularly as an alternative for Molybdate Red pigments in paints. Systems containing P.R.251 are only fast to bleeding up to 80–100 °C. The pigment is frequently found in long-oil alkyd resin paints (house paints), in medium-oil alkyd resin systems (air drying industrial paints, including systems that are dried at elevated temperature up to about 80 °C, such as automotive refinishes), as well as in emulsion paints. P.R.251 exhibits excellent lightfastness and weatherfastness.
3.7.2 Hydroxyanthraquinone Pigments
The class of hydroxyanthraquinone pigments consists of two different groups of compounds: metal complexes of hydroxyanthraquinones on the one hand and metal salts of hydroxyanthraquinone sulfonic acids on the other hand. Some of the products are metal chelates.
The first group includes 1,2-dihydroxyanthraquinone, commonly known as alizarin, 1,4-dihydroxyanthraquinone (quinizarin), and 1,2,4-trihydroxyanthraquinone (purpurin). Alizarin in particular has been known and appreciated for thousands of years in the form of its ‘lake’, that is, the coordination complex of 1,2-hydroxyanthraquinone with aluminium and calcium, 70 (Madder Lake, Turkey Red). The chemical structure of 70 was first described in 1963 [166]:

Aluminium and calcium are equally important constituents of other hydroxyanthraquinone compounds, although iron salts are also known.
The calcium lake of 1,2-dihydroxyanthraquinone is marketed as Pigment Red 83, C.I. 58000:1. It is produced commercially by treating a slightly basic alizarin solution with aqueous calcium chloride:

Scheme 3.14 Molecular structure of P.R.83.
All of these compounds were originally precipitated onto aluminium oxide hydrate, which served as a carrier. This applies accordingly to hydroxyanthraquinone sulfonic acids.
Both groups have lost most of their commercial importance as pigments.
There is one derivative of hydroxyanthraquinone sulfonic acid that is used to some extent. This is the reddish violet Pigment Violet 5:1, C.I. 58055:1, which has the basic structure 71:

Scheme 3.15 Molecular structures of P.V.5, P.V.5:1, P.V.6, P.V.6:1, P.V.7 and P.V.7:1.
This compound is obtained from phthalic acid and p-chlorophenol with sulfuric acid in the presence of boric acid. The intermediate product is quinizarin, which is sulfonated in oleum or with sodium hydrogen sulfite and oxidants to form 71:

3.7.2.1 Commercially Available Hydroxyanthraquinone Pigments
3.7.2.1.1 Pigment Red 83
P.R.83 (formula see Scheme 3.14), listed under Constitution Number 58000:1, continues to be used only in the USA. The pigment affords brilliant, bluish shades of red. Traces of iron as an impurity adversely affect the full shades and shift the colour towards duller and bluer shades. The pigment is not fast to common organic solvents, especially to esters and ketones. It therefore lacks stability to overcoating. Its lightfastness, particularly in tint, is poor. P.R.83 is used in paints for toys, in packaging printing inks, especially for soap and butter, and in artists' colours.
3.7.2.1.2 Pigment Violet 5:1
Commercial interest in P.V.5:1, Constitution No. 58055:1 (Al3+ complex of 71, see above), has declined considerably. The pigment continues to be used in industrial paints, especially throughout the USA. Its full shade is a brilliant, deep bluish maroon. In white reductions, the pigment produces clean, reddish violet shades. It lacks tinctorial strength and the coatings are fast to neither acid nor alkali. P.V.5:1 is also not very lightfast, which practically precludes its use in products for exterior application, particularly in reduced shades.
P.V.5:1 is used to a certain extent in PVC. Plasticized PVC systems are fast to bleeding and reasonably lightfast in full shades. Addition of TiO2, however, markedly affects its lightfastness. The pigment is heat stable up to 170 °C.
3.7.3 Heterocyclic Anthraquinone Pigments
As a group, these pigments include compounds whose heterocyclic system is formally derived from the anthraquinone nucleus.
3.7.3.1 Anthrapyrimidine Pigments
1,9-Anthrapyrimidine (72), which is not used commercially, provides the parent structure for an important yellow pigment:

Anthrapyrimidine and its substituted derivatives are obtained by condensation of 1-aminoanthraquinone (or its derivatives) with formamide or aqueous formaldehyde/ammonia in the presence of an oxidant, such as ammonium vanadate or m-nitrobenzosulfonic acid. A newly developed, simpler route proceeds via formamidinium chloride, which is prepared from 1-aminoanthraquinone with dimethylformamide and thionyl chloride or phosphorus oxychloride. Cyclization in a solvent in the presence of ammonium acetate affords the desired product:
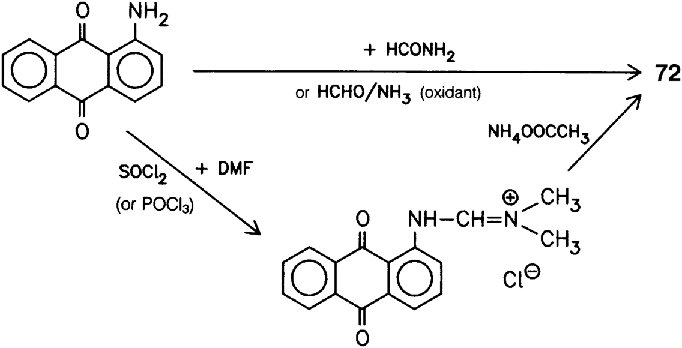
Vat Yellow 20, a product long known and patented as early as 1935, has the following chemical constitution, derived from anthrapyrimidine:
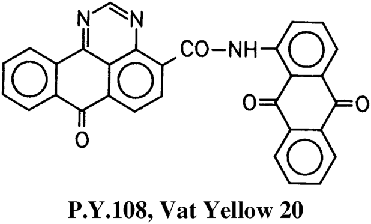
Scheme 3.16 Molecular structure of P.Y.108.
The compound has gained commercial recognition primarily as an organic pigment (Pigment Yellow 108, C.I. 68420).
P.Y.108 is obtained under condensation from 1-aminoanthraquinone, 1,9-anthrapyrimidine-2-carboxylic acid and a chlorinating agent, or directly with 1,9-anthrapyrimidine-2-carboxylic acid chloride in an organic solvent in the presence of an acid trap.
Thus, P.Y.108 is commercially produced by heating 1,9-anthrapyrimidine-2-carboxylic acid with 1-aminoanthraquinone and thionyl chloride in a high-boiling solvent, such as o-dichlorobenzene or nitrobenzene, to 140–160 °C. The product is separated, washed with methanol, and residual solvent removed by steam distillation. The aqueous suspension is then boiled down with sodium hypochlorite solution.
Finer particle sizes are obtained if 1,9-anthrapyrimidine-2-carboxylic acid chloride is condensed with 1-aminoanthraquinone in a dipolar aprotic solvent (such as N-methylpyrrolidone) at a temperature between 70 and 110 °C. The reaction may be accelerated by using a proton acceptor such as triethylamine or tert-butanol, which reacts with hydrochloric acid. A particularly useful route proceeds via the acid chloride, which is prepared by reacting anthrapyrimidine carboxylic acid with thionyl chloride at 40 °C. A condensation reaction follows without separation of the intermediate [167].
3.7.3.1.1 Properties and Application of Pigment Yellow 108
Anthrapyrimidine Yellow (formula see Scheme 3.16) is primarily applied in paints. In lighter tints, the pigment provides a medium, dull yellow colour, while full shades and similarly deep shades are distinctly redder and even duller than tints. P.Y.108 is not fast to the solvents commonly used in paints. It bleeds more or less into solvents such as aromatic and aliphatic hydrocarbons, alcohols, ketones and esters (Section 1.6.2.1). P.Y.108 accordingly lacks entire fastness to overcoating. As a colourant for coatings, it is heat stable up to 160 °C. P.Y.108 is both very lightfast and weatherfast in lighter tints, but its fastness to light and weather deteriorates rapidly as more white pigment is added. Some darkening is observed. P.Y.108 is therefore used particularly in medium to very light shades, as a shading pigment, and as a source of cream colours. For a long time, such P.Y.108 systems were considered the most weatherfast products within the medium yellow portion of the spectrum.
P.Y.108 is applied in various types of industrial finishes, especially in original automotive (OEM) and in automotive refinishes. It is also recommended for metallic finishes, although it is much less weatherfast in such systems. The pigment tends to seed, that is, it forms specks upon storage. The mechanism behind this phenomenon remains to be elucidated. In addition, P.Y.108 also lends colour to emulsion paints, in which it is durable enough to satisfy the requirements for exterior paints based on synthetic resin dispersions. It is also fast to acids, alkali and plaster.
The printing inks industry utilizes P.Y.108 only in special-purpose media, including packaging inks, metal decorating inks, posters and similar applications. The prints are comparatively dull, but very lightfast. They may safely be sterilized.
3.7.3.2 Indanthrone and Flavanthrone Pigments
In 1901, R. Bohn synthesized indanthrone and flavanthrone. Both compounds are thus amongst the oldest synthetic vat dyes known.
3.7.3.2.1 Indanthrone
Vat dyes of the highest quality have derived their name from indanthrone, a blue compound, which was originally known as Indanthrene Blue. The compound has long been used as a pigment and is registered in the Colour Index as Pigment Blue 60, C.I. 69800 (73):

Scheme 3.17 Molecular structure of P.B.60.
The primary synthetic route proceeds via oxidative dimerization of 2-aminoanthraquinone in the presence of an alkali hydroxide. 2-Aminoanthraquinone, for instance, is fused with potassium hydroxide/sodium hydroxide at 220–225 °C in the presence of sodium nitrate as an oxidant. New techniques involve air oxidation of 1-aminoanthraquinone at 210–220 °C in a potassium phenolate/sodium acetate melt or in the presence of small amounts of dimethyl sulfoxide. A certain amount of water that is formed during the reaction may be removed by distillation to improve both efficiency and yield.
One out of several possible options for the synthesis of indanthrone from 2-aminoanthraquinone is illustrated in the following scheme:

The reactions involve various preliminary equilibria, which open up several different pathways. Therefore, the reaction conditions during indanthrone synthesis must be followed exactly [168, 169].
The pigment form [170] is obtained from the leuco form, which in turn is prepared by oxidation with alkali fusion, followed by treatment with sodium hydrogen sulfite solution or sodium dithionite (vatting).
Heating the leuco form in an aqueous alkaline medium in the presence of air, possibly also in the presence of a surfactant, affords the pigment form. Oxidation may also be achieved with sodium m-nitrobenzenesulfonate. Another alternative is to precipitate the vat acid from the salt of the leuco form and to subsequently oxidize the product. It is also possible to mill the suspension of the leuco form together with aqueous sodium hydroxide/sodium dithionite in air, using a pearl mill.
Other methods start from crude indanthrone, which is dissolved in sulfuric acid or oleum (sometimes mixed with organic solvents). The product may be precipitated with water. It is more advantageous to separate the precipitated crude pigment, to reslurry it in water, and to heat the resulting aqueous suspension in the presence of a cationic surfactant. Subsequent treatment of the sulfuric acid solution with nitric acid, manganese dioxide or chromium trioxide is followed by transfer into a solution containing sodium sulfite or iron(II) sulfate.
Kneading or milling the crude indanthrone in the presence of finishing agents, such as polyols, or milling it with salt also affords a product that provides useful pigment properties.
In the past, not only unsubstituted indanthrone but also some chloro derivatives were commercially available. 3,3′-Dichloroindanthrone, also known as Pigment Blue 64, C.I. 69825, has had a certain impact on the market:

Scheme 3.18 Molecular structure of P.B.64.
No current manufacturers are known of this and other indanthrone derivatives that are preferably chlorinated in α-position (4,4′-, 5,5′- or 8,8′-) and used as pigments.
3.7.3.2.2 Flavanthrone
Flavanthrone has also long been used as a vat dye. It gained recognition as a pigment when increasingly lightfast and durable paints were required.
The yellow pigment is registered in the Colour Index as Pigment Yellow 24, C.I. 70600. It has the structure 76:

Scheme 3.19 Molecular structure of P.Y.24 (temporarily known as P.Y.112).
The synthetic route involves treating 1-chloro-2-aminoanthraquinone with phthalic anhydride (PA), which initially affords 1-chloro-2-phthalimidoanthraquinone (77). Subsequent Ullmann reaction with copper powder in refluxing trichlorobenzene yields 2,2′-diphthalimido-1,1′-dianthraquinonyl (78) through dimerization. This product is cyclized with 5% aqueous sodium hydroxide solution at 100 °C, cleaving phthalic acid units, to yield 76:

This three-step synthesis may be performed in one step by heating 1-halogeno-2-aminoanthraquinone with copper in a highly polar aprotic solvent [171].
An older, equally interesting industrial route involves condensing 2-aminoanthraquinone in nitrobenzene in the presence of antimony pentachloride or titanium tetrachloride. Complex 79 prevents any undesirable formation of anthrimide (80):

The comparatively low yield, together with the high price of antimony pentachloride, make this process economically unattractive.
A more recent method starts from 2,2′-diacetylamino-1,1′-dianthraquinonyl, which is cyclized in a phase-transfer reaction in chlorobenzene using tetrabutylammonium bromide and a 30% aqueous sodium hydroxide solution [172]:

Flavanthrone, like indanthrone, must be extremely pure in order to develop useful pigment properties. Subsequent finishing converts the thus-prepared material into an appropriate product for use in paints or plastics.
The crude product may be purified by one of the following methods:
- dissolving the material in concentrated sulfuric acid at 50–70 °C and hydrolysing the thus prepared sulfate with water;
- converting the crude product into its leuco form, separating the intermediate, and reoxidizing the compound to form pure flavanthrone;
- extracting the crude product with a dipolar aprotic solvent, such as dimethylformamide or dimethyl sulfoxide.
Various milling techniques are available to develop the desired pigment properties, performing the finishing process either in an aqueous suspension or in the presence of organic solvents or milling agents. A pigment prepared by this route typically evolves in an opaque, reddish yellow form.
A product that provides the same shade but higher transparency is obtained by converting crude flavanthrone into its leuco form (for instance with sodium dithionite/aqueous sodium hydroxide). The resulting intermediate material is separated and resuspended in water. It undergoes facile reoxidation through shearing forces and/or in the presence of surfactants, affording the pigment [173].
The crude pigment may also be treated with an aromatic sulfonic acid (such as toluene sulfonic acid, xylene sulfonic acids, m-nitrobenzene sulfonic acid) in sulfuric acid or with nitric acid at 80 °C to yield a somewhat redder yellow transparent modification of flavanthrone [174].
3.7.3.2.3 Polymorphism and Crystal Structures of Indanthrone and Flavanthrone
Indanthrone exists in four polymorphic forms [175]. From the investigations of the British Intelligence Office and the corresponding US authorities in Germany after the Second World War it is known that these polymorphic forms were investigated by X-ray powder diffraction at the BASF company as early as in 1934 [176, 177], that is, only 18 years after the diffraction of X-rays on powders was discovered by Debye and Scherrer in 1916.
The α- and the β-modification afford greenish and reddish blue shades, respectively, while the γ-form provides reddish hues. The δ-modification is a reddish blue pigment of high transparency, strong weather resistance and superior dispersibility [178]. Being the most stable thermodynamically, the α-form is most suitable for use as a pigment. It affords reddish and greenish blue grades. The more greenish type is prepared by precipitating a crude indanthrone solution in sulfuric acid, or it is obtained from the leuco form with a surfactant by air oxidation. The only commercially available reddish form is also obtained through air oxidation, although it is necessary to simultaneously apply shearing forces [179, 180].
The indanthrone and flavanthrone molecules have a similar molecular shape. The crystal structures of the α-phase of indanthrone and of flavanthrone are similar as well (Figure 3.61) [181, 182]. The NH groups of indanthrone forms intramolecular hydrogen bonds. In both compounds the molecules are connected by van der Waals and Coulomb interactions only. The molecules crystallize in stacks, as it is frequently observed for organic pigments. Molecules from neighbouring stacks arrange in a herringbone pattern (Figures 3.61 and 3.62).

Figure 3.61 Crystal structures of (a) indanthrone (α-phase) and (b) flavanthrone. View along the stacking direction.

Figure 3.62 Crystal structures of (a) indanthrone (α-phase) and (b) flavanthrone. View perpendicular to the stacking direction, showing the herringbone arrangement of the molecules.
The density of indanthrone and flavanthrone is quite high (1.58 g cm−3), which results in favourable lattice energies and good fastness properties. Figure 3.63 shows the space-filling, dense packing of molecules in flavanthrone.

Figure 3.63 Crystal structure of flavanthrone. The molecules form a very dense packing.
3.7.3.2.4 Properties and Application of Indanthrone (Pigment Blue 60)
Indanthrone (formula see Scheme 3.17) demonstrates excellent weatherfastness. Most grades of P.B.60, like other commercial vat type pigments, provide good transparency. They are primarily applied in automotive finishes, especially in metallic finishes. In these media, P.B.60 is even more weatherfast than copper phthalocyanine pigments, especially in light tints. This makes P.B.60 particularly attractive in areas where copper phthalocyanine pigments are not weatherfast enough to satisfy the application requirements. P.B.60 is highly durable even in light white reductions. Its shade is noticeably redder and duller than α-Copper Phthalocyanine Blue. Likewise, P.B.60 is also duller than the similarly redder ɛ-Copper Phthalocyanine Blue. Although a pigment with high tinctorial strength, P.B.60 is weaker than α-Copper Phthalocyanine Blue types. P.B.60 shows very good fastness to organic solvents, including alcohols, esters, and ketones, aromatic and aliphatic hydrocarbons, and various types of plasticizers. Incorporated in baked enamels, the pigment is fast to overcoating. P.B.60 is also fast to acids and alkali and heat stable up to 180 °C. The pigment is not only used in automotive finishes but also in general industrial paints, wherever the high demands on pigment fastness properties or the very reddish shade warrant its use. Notably, however, a very similar shade is accessible through shading of the more brilliant α-Copper Phthalocyanine Blue with Dioxazine Violet. The cleanness of the product may be reduced if needed. The resulting mixture, however, is less weatherfast in light tints than Indanthrone Blue, especially in metallic finishes. Adding suitable UV absorbers eliminates the difference in weatherfastness.
In addition, P.B.60 is employed in plastics wherever its shade is required, or for its excellent fastness properties. The pigment has the advantage of being highly heat stable. Dispersed in polyolefins, it is fast up to 300 °C for 5 min. The difference in colour between full shades and 1/3 SD HDPE samples at 300 °C as opposed to 200 °C is only approximately 1.5 CIELAB units. 1/25 SD specimens are thermally stable up to 280 °C. Indanthrone Blue exhibits average tinctorial strength: 0.15% pigment is necessary to produce 1/3 SD colourations with 1% TiO2. The same result is achieved with as little as approximately 0.08% α-Copper Phthalocyanine Blue. P.B.60 does not nucleate, that is, it does not affect the shrinkage of injection-moulded plastic articles to a noticeable extent. As in PVC, P.B.60 is frequently used in these materials as an alternative to phthalocyanine blue pigments for its redder shade. It is almost entirely fast to bleeding in plasticized PVC. The pigment shows excellent lightfastness: 1/3 SD colourations equal step 8 on the Blue Scale. Likewise, P.B.60 is also highly weatherfast, fast enough to be recommended for long-term exposure. This pigment is a valuable colourant for a wide variety of plastic media. Used in PP spin dyeing, for instance, P.B.60 exhibits greater fastness than copper phthalocyanine pigments. It is a suitable pigment for the colouration of rubber and other elastomers. Polyamide is an unsuitable medium for P.B.60.
Printing inks made of P.B.60 are used to print banknotes.
Commonly, the α-crystal modification of P.B.60 is commercially most often used. Some years ago the δ-crystal modification of P.B.60 was developed. It has the characteristics of the α-crystal pigment, but is significantly redder in shade. Blending the δ-crystal form with P.V.19 results in very red shades of blue, which have higher chroma compared to blends with the α-crystal form [93].
3.7.3.2.5 Properties and Application of Flavanthrone (Pigment Yellow 24)
Flavanthrone Yellow (formula see Scheme 3.19), together with its chemical structure, is listed in the Colour Index under Constitution No. 70600. It was temporarily known as Pigment Yellow 112, but now it is exclusively referred to as Pigment Yellow 24. For some time sales products of P.Y.24 have no longer been listed in the catalogues of manufacturers, but the grades are still available on the market.
The commercially available types are reddish yellow. They are speciality products for use in paints. In addition, P.Y.24 is also employed for polyacrylonitrile spin dyeing. The pigment equals the considerably more greenish and distinctly weaker Anthrapyrimidine Yellow in terms of fastness to organic solvents and chemicals. P.Y.24 is almost entirely fast to overcoating. Full shades and similarly deep shades darken considerably upon exposure to light and weather. This effect, however, is not noticeable in very light tints; corresponding samples show very good lightfastness and weatherfastness. P.Y.24 is, however, not quite as durable as anthrapyrimidine yellow pigments, such as P.Y.108.
High transparency makes P.Y.24 a valuable pigment for metallic finishes. It is used in relatively light shades, typically at a ratio of one part of colour pigment to three parts of aluminium pigment. Thus-prepared systems demonstrate excellent weatherfastness. Flavanthrone Yellow, like P.Y.108, tends to seed (Section 3.7.3.1). The pigment is heat stable up to 200 °C and thus satisfies all possible heat stability requirements in this area. Flavanthrone Yellow is used in various industrial paints, especially in automobile OEM finishes and in automotive refinishes.
Although highly reduced P.Y.24 plastic systems are also very lightfast, such products are used only to a limited extent. P.Y.24 shows average tinctorial strength. 1/3 SD colourations in HDPE (1% TiO2), for instance, are formulated at approximately 0.25% pigment. Such products may be safely exposed to temperatures up to 270 °C, while addition of TiO2 (1/25 SD) reduces the heat stability of the pigmented systems to 230 °C. The tinctorial strength increases at higher temperature and the colour turns greener, a phenomenon that points to pigment dissolution. Polyacrylonitrile spin dyed fibres are lightfast and weatherfast enough to satisfy stringent requirements, which qualifies the pigment for use in canvasses and similar media. In this respect, P.Y.24 is something of an exception amongst organic yellow pigments.
P.Y.24 is also sold for use in other areas in which high lightfastness is a prime concern. It is thus used in media such as solvent-based wood stains and in artists' colours.
3.7.4 Polycarbocyclic Anthraquinone Pigments
This class includes polycarbocyclic compounds that are at least formally derived from the anthraquinone structure. The products are considered members of the higher condensed carbocyclic quinone series, which even in the absence of additional substituents provide yellow to red shades. Halogenation is frequently found to afford cleaner shades and improved fastness properties. Heading the list of such derivatives are pyranthrone, anthanthrone and isoviolanthrone pigments.
3.7.4.1 Pyranthrone Pigments
Pyranthrone pigments are related to the pyranthrone structure (81), which is formally derived from flavanthrone (Section 3.7.3.2) in that the nitrogen atoms are replaced by CH groups:
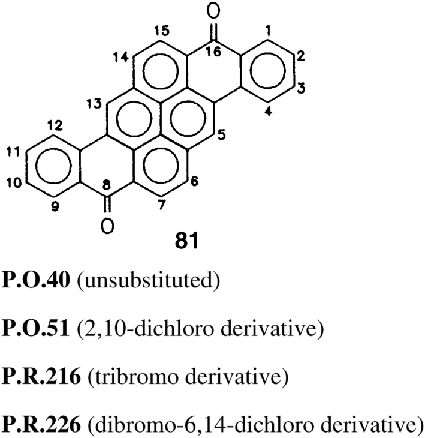
Scheme 3.20 Molecular structures of P.O.40, P.O.51, P.R.216, and P.R.226.
Manufacture of the unsubstituted compound 81 (Pigment Orange 40, C.I. 59700) has been discontinued. Other commercially available pyranthrone pigments include bromo, chloro or bromo/chloro derivatives of the parent structure.
Pyranthrone is commonly prepared by Ullmann reaction of 1-chloro-2-methylanthraquinone (82), followed by double ring closure.
1-Chloro-2-methylanthraquinone is treated with copper powder, pyridine and dry sodium carbonate in o-dichlorobenzene at 150–180 °C to form 2,2′-dimethyl-1,1′-dianthraquinonyl (83). Subsequent condensation is achieved after several hours of boiling in a sodium hydroxide/isobutanol solution at 105 °C, which affords the leuco form. The product is then oxidized to form pyranthrone by blowing air through the reaction mixture:

Heating 83 with aqueous sodium hydroxide in diethylene glycol monomethylether or with alkali acetate, a reaction that may also be performed in other polar organic solvents, such as dimethylformamide, N-methylpyrrolidone, or dimethylacetamide at 150–210 °C, also provides pyranthrone. As an alternative, 83 may be cyclized to form pyranthrone in a two-phase reaction, carried out in the presence of quaternary ammonium salts in a phase-transfer system consisting an aqueous and an organic phase [171].
Halogenated pyranthrones may be obtained by two different routes. One route proceeds via ring closure of halogenated 2,2′-dialkyl-1,1′-dianthraquinonyl, in accordance with the synthesis of pyranthrone. The same end may be accomplished by halogenation of ready-made unsubstituted pyranthrone.
Compound 83, for instance, may be chlorinated in o-dichlorobenzene, followed by ring closure through phase-transfer reaction with aqueous sodium hydroxide/water and dichlorobenzene. 3,3′-Dichloro-2,2′-dimethyl-1,1′-dianthraquinonyl is also accessible through ring closure by traditional alkaline condensation or by adding polar solvents in the presence of sodium acetate at 110–150 °C [183].
3,3′-Dichloro-2,2′-dimethyl-1,1′-dianthraquinonyl can be synthesized by Friedel–Crafts reaction from phthalic anhydride and 2,6-dichlorotoluene. Subsequent cyclization of the resulting substituted benzoylbenzoic acid 84 in sulfuric acid affords the corresponding anthraquinone derivative 85, which is dimerized by an Ullmann reaction:

Pyranthrone may be halogenated, for instance, in chlorosulfonic acid in the presence of small amounts of sulfur, iodine or antimony as a catalyst. This procedure necessitates intermediate separation and purification of pyranthrone after manufacture, because the products, unless purified, fail to furnish the solvent fastness that is characteristic of a typical pigment.
While 3,3′-dichloro-2,2′-dimethyl-1,1′-dianthraquinonyl is easily converted into well-defined 6,14-dichloropyranthrone, direct halogenation affords changing amounts of chloro and bromo derivatives with undefined substitution patterns on the pyranthrone ring. Depending on the amount of added halogen, an average of two to four halogen atoms per molecule is expected.
Bromination of 6,14-dichloropyranthrone in chlorosulfonic acid in the presence of a catalyst provides 0.1–2.2 bromine atoms per molecule, depending on the reaction conditions. The exact outcome is controlled not only by the choice of solvent but also by the type of catalyst and the amount of bromine, as well as by the reaction time and reaction temperature.
The halogenated pyranthrones dissolved in chlorosulfonic acid may be isolated by precipitation into ice water. Residual acid is removed by washing. The thus-prepared products, such as 6,14-dichloropyranthrone, do not require further treatment before bromination. An opaque version of 6,14-dichloropyranthrone as well as transparent types of chlorobromopyranthrones are accessible by initially dissolving the crude pigment in sulfuric acid, reprecipitating it with ice/water, and separating the product. The aqueous presscake is then treated with C4–C10-alkanols or alkanones, or with C6–C8-cycloalkanols or alkanones [184].
An opaque form of pyranthrone may also be prepared by treating the corresponding pigment in a polar organic solvent (such as isobutanol) at elevated temperature in the presence of some (0.5–10%) halogenated anthraquinone or another anthraquinone derivative.
While 7,15- and 3,11-dichloropyranthrone demonstrate such poor solvent fastness as to be unsuitable for use as pigments, 6,14-dichloropyranthrone possesses excellent solvent resistance.
In addition to 6,14-dibromopyranthrone, some mixed halogenated pyranthrone derivatives exhibit equally useful pigment properties.
3.7.4.1.1 Commercially Available Pyranthrone Pigments and Their Application
Pyranthrone and some of the halogenated pyranthrone types had been offered to the market. The producer has ceased their production and does no longer lists them in the sales catalogues. The pigments, however, are still available on the market, giving reason to further characterize their application and fastness properties in this book.
3.7.4.1.1.1 Pigment Orange 40
P.O.40, unsubstituted pyranthrone (see Scheme 3.20), is registered under Constitution Number 59700. Its production has been discontinued. The pigment used to be employed in spin dyed viscose rayon and viscose cellulose. The resulting products demonstrate good lightfastness. 1/1 to 1/25 SD specimens equal step 6–7 to step 6 on the Blue Scale. The pigment exhibits average tinctorial strength. Its textile fastnesses are excellent. As a result of its chemical constitution, P.O.40 is not vat resistant, that is, it is only moderately fast to an alkaline sodium dithionite solution (60 °C for 30 min). Spin dyed fibres undergo noticeable colour change under such conditions – they bleed and distinctly colour white staple fibre and cotton.
3.7.4.1.1.2 Pigment Orange 51
P.O.51, which is the 2,10-dichloropyranthrone derivative (Scheme 3.20), affords a medium orange shade. It is not only slightly redder than the halogen-free variety, but also exhibits higher cleanness and is tinctorially stronger in tints. The pigment is used especially to provide reddish shades of yellow. P.O.51 provides good fastness to organic solvents, it may safely be overcoated under normal conditions if incorporated in baking enamels. Heat stable up to more than 200 °C, P.O.51 satisfies practically all possible requirements for use in this area. Its full shade and similarly deep shades, down to approximately 1/9 SD, show excellent lightfastness and weatherfastness. The commercial version possesses only poor transparency, despite its high specific surface area of about 60 m2 g−1. This brand is also recommended for use in metallic shades and is used extensively in two-coat metallic finishes. Although highly weatherfast, medium colours do not perform as well as the other halogenated, redder pyranthrone pigments.
3.7.4.1.1.3 Pigment Red 216
P.R.216, Constitution Number 59710, a tribromopyranthrone derivative (Scheme 3.20), is distinctly bluer than P.R.226, and its shade is comparatively dull. As a result of a high specific surface area of about 80 m2 g−1, the commercial version is highly transparent. The pigment is specifically recommended for use in metallic finishes. P.R.216, like P.R.226, is frequently used in two-coat metallic finishes for automobiles, especially if very little coloured pigment is applied. Combination with UV absorbants in the top coat is also common. Pigment durability in medium tints, like 1/3 SD, is good. However, the weatherfastness decreases rapidly as more TiO2 is added. This effect is more noticeable in P.R.216 than in P.R.226 (see below). P.R.216 is also employed in pigment combinations with coloured inorganic pigments, especially with Molybdate Red derivatives. It shows good fastness to solvents and performs even better in this respect than P.R.226. This is also true for the fastness to overcoating, although baking temperatures above 140 °C should be avoided.
P.R.216 lends colour to all types of industrial paints, it is heat stable up to 200 °C. Like other pyranthrone pigments, it is suitable for use in unsaturated polyester systems, in which it is resistant to peroxides.
P.R.216 demonstrates excellent lightfastness in plastics, especially in PVC. 1/3 SD plasticized PVC systems (5% TiO2) equal step 8 on the Blue Scale for lightfastness. Pigment weatherfastness is similarly excellent, but fails to meet the standards for long-term exposure. P.R.216 is not entirely fast to bleeding, and its tinctorial strength is poor: 1/3 SD (5% TiO2) is formulated at 1.85% pigment. 1/3 SD colourations in HDPE with 1% TiO2 withstand exposure to 250 °C. In terms of lightfastness, these specimens equal step 6 on the Blue Scale.
3.7.4.1.1.4 Pigment Red 226
Pigment Red 226 (Scheme 3.20) is a dibromo-6,14-dichloro-pyranthrone. It provides a medium, somewhat dull red shade, which is considerably more bluish than that of P.O.51. The shade, although noticeably duller, corresponds to that of the less fast toluidine red pigments. P.R.226 is distinctly yellower than P.R.216. Despite the fact that P.R.226 demonstrates good fastness to solvents, it is much less resistant than other members of its class. The difference is most noticeable in toluene and xylene. P.R.226 is fast to acids and alkali. Like P.O.51, it is thermally stable up to more than 200 °C, even in long-term exposure for several weeks. The commercial grade is more transparent than P.O.51 and provides excellent lightfastness and weatherfastness. P.R.226 is a special-purpose type that is used for metallic finishes. It is a suitable pigment for automotive finishes, especially for two-coat metallic finishes, quite often in combination with UV absorbants in the clean top coat.
3.7.4.2 Anthanthrone Pigments
Anthanthrone pigments are characterized by the basic structure 86:

The unsubstituted ring system, although exhibiting an orange shade, apart from other deficiencies is tinctorially not strong enough to stimulate interest. Only halogenated derivatives have gained some interest throughout the pigment industry.
Anthanthrone is synthesized from naphthostyril (87), which is saponified to form 1-aminonaphthalene-8-carboxylic acid (88). Naphthostyril itself is prepared from 1-naphthylamine with phosgene in the presence of dry aluminium chloride.
Diazotizing 88 and boiling the solution in the presence of copper powder affords 1,1′- dinaphthyl-8,8′-dicarboxylic acid (89), which is cyclized with aluminium chloride, but preferably with concentrated sulfuric acid at 30–40 °C, to produce anthanthrone (86):

The Friedel–Crafts reaction, which proceeds via electrophilic aromatic substitution, as illustrated in the following scheme, is unique to the manufacture of anthanthrone pigments. Most other polycyclic anthraquinone pigments are synthesized via nucleophilic ring closure.

4,10-Dibromoanthanthrone (90) is registered as Pigment Red 168, C.I. 59300. First synthesized as early as 1913 as a vat dye, this compound is the commercially most interesting halogenated anthanthrone derivative:

Scheme 3.21 Molecular structure of P.R.168.
Compound 90 may be prepared directly from 86 without intermediate isolation of 86 by treating the dicarboxylic acid 89 with monohydrate or with concentrated sulfuric acid at 35 °C, followed by bromination in the presence of iodine as a catalyst.
P.R.168 exists in two polymorphic forms [185]. The metastable β-polymorph is formed upon synthesis. This orange product is mainly used as a vat dye. Finishing, for example by recrystallization from sulfuric acid, leads to the thermodynamically stable scarlet α-phase, which is used as a pigment.
In the α-phase of P.R.168 the molecules adopt a ‘brick’ packing, as it is also found in the β-phase of P.R.224 (Figure 3.64). While the molecules arrange in a herringbone arrangement in P.R.168, they form slightly wavy layers in P.R.224 (Figure 3.65). Similarly as in P.R.224, the molecular packing is extremely efficient and dense (Figure 3.66).
Figure 3.64 Comparison of the crystal structures of (a) P.R.168 (α-phase) and (b) P.R.224 (β-phase); view along the stacking direction.

Figure 3.65 Comparison of the crystal structures of (a) P.R.168 (α-phase) and (b) P.R.224 (β-phase); view perpendicular to the stacking direction.

Figure 3.66 Perfect space filling in the crystal structure of P.R.168 (α-phase).
In contrast to the reddish orange colour of P.R.168, which is referred to as scarlet, the yellower 4,10-dichloroanthanthrone is of no commercial interest as a pigment and is therefore no longer produced.
3.7.4.2.1 Commercially Available Anthanthrone Pigments
3.7.4.2.1.1 Pigment Red 168
Pigment Red 168 dibromoanthanthrone, is a vat type pigment that demonstrates excellent fastness properties, a feature that is an asset in high grade paints. It provides a clean yellowish shade of scarlet, somewhere between that of P.O.43, a naphthalene tetracarboxylic acid derivative, and those of yellowish perylene tetracarboxylic acid type pigments.
P.R.168 is entirely or almost entirely resistant to most of the organic solvents commonly found in typical binder systems. P.R.168, like other vat type pigments, is not completely fast to overcoating in oven drying systems that are baked at 120–160 °C. The degree of bleeding depends on the individual system. The pigment is thermally stable up to 180 °C. It is one of the most lightfast and weatherfast organic pigments known. Excellent performance standards make it a suitable candidate for all types of coatings and paints, even at very low pigment concentrations. P.R.168 is used both as a shading pigment and in mixed systems. It exhibits comparatively low tinctorial strength. The commercial products, like those of other vat type pigments, are more or less transparent, which makes them useful products for metallic finishes. P.R.168 is used, for instance, in one- and two-coat automotive finishes. Moreover, it is weatherfast enough to be used in low concentrations. In such systems, P.R.168 is also employed in combination with highly fast, typically reddish yellow pigments to produce shades of bronze and copper. The pigment is equally useful in conjunction with more reddish organic pigments, such as with perylenetetracarboxylic acid pigments. High weatherfastness makes it a suitable candidate for use in architectural paints and in emulsion paints, including those targeted for outdoor use. The fact that P.R.168 is only used in very light tint allows its comparatively high price in this market. Specialized pigment preparations are also available for these purposes. P.R.168 is fast to alkali and plaster. The pigment is also recommended for use in coil coating, including PVC coatings (Section 1.8.2.2).
P.R.168 is found to a lesser extent in printing inks and plastics. The printing ink industry utilizes P.R.168 to produce special-purpose printing inks, which may be applied to substrates such as posters or metal deco prints. The pigment demonstrates equally excellent fastness in these materials. 1/1 SD systems equal step 8 on the Blue Scale for lightfastness, while 1/3 to 1/25 SD formulations match step 7. The prints are resistant to common organic solvents and chemicals. The pigment is thermally stable up to 220 °C for 10 min, and its prints may safely be sterilized.
3.7.4.2.1.2 Pigment Orange 77
The pigment is listed in the Colour Index under Constitution Number 59105:

Scheme 3.22 Molecular structure of P.O.77.
It is described as bright yellowish orange and can be used for aqueous flexo inks and in the plastics application in dibutyl phthalate paste as plasticiser for PVC. Dyeing of technical leathers is as well described.
3.7.4.3 Isoviolanthrone Pigments
Isoviolanthrone (91) is an highly anellated polycyclic quinone system. It is derived from the chemical structure of isodibenzanthrone, which may be visualized as being obtained by unsymmetrical condensation of two benzanthrone (92) molecules. The compound isoviolanthrone itself affords an intense blue shade.

Isoviolanthrone is prepared from benzanthrone, which is synthesized from anthraquinone. Anthraquinone is reacted with glycerine and sulfuric acid in the presence of a reducing agent such as iron. In this reaction, anthraquinone is initially reduced to anthrone (93), which is condensed with acrolein. Acrolein, on the other hand, is an intermediate of the reaction between glycerine and sulfuric acid. The synthesis proceeds by oxidative cyclization with sulfuric acid. The resulting benzanthrone (92) is halogenated in sulfuric acid or in chlorosulfonic acid in the presence of a catalyst (sulfur, iodine, or iron) to afford 3-chloro or 3-bromobenzanthrone (93a). Subsequent treatment with sodium disulfide at 135 to 150°C under pressure leads to 3,3′-dibenzanthronyl sulfide (93b). Finally, treating boiling 93b with potassium hydroxide in alcohol (ethanol or isobutanol) leads to isoviolanthrone (91).
In contrast to earlier methods which involved an alcoholic potassium hydroxide fusion of 3-chloro-benzanthrone, this route affords pure isoviolanthrone, free of isomers.

Isoviolanthrone was first synthesized in 1907 and has long been known as a vat dye. However, in contrast to its halogenated derivatives, unsubstituted isoviolanthrone has failed to gain technical recognition as a pigment.
Pigment Violet 31, C.I. 60010, a dichloroisoviolanthrone (94), is commercially available. A monobromo derivative is registered as P.V.33.
Dichloroisoviolanthrone is prepared by chlorination of isoviolanthrone with sulfuryl chloride in nitrobenzene. Chlorine substitution occurs at positions 6 and 15:

Scheme 3.23 Molecular structure of P.V.31.
Compounds containing 12–17% bromine (and up to 1% chlorine) are referred to as bromoisoviolanthrone. The product has been discontinued in the USA. It is obtained by bromination of isoviolanthrone at 80 °C in chlorosulfonic acid, using iodine as a catalyst. Two crystal modifications are obtained: the α-modification is produced by treating crude isoviolanthrone in 90% sulfuric acid and discharging it into an aqueous dispersion solution. The β-modification evolves if the aqueous α-paste is heated in the presence of N-methylpyrrolidone and a surfactant, or generally by stirring the α-modification with organic solvents [186].
A higher quality α-modification with enhanced tinctorial strength and transparency is prepared from the leuco form of bromoisoviolanthrone. This intermediate in turn is manufactured by vatting crude bromoisoviolanthrone with sodium dithionite/aqueous sodium hydroxide. The product is separated and oxidized in an aqueous alkaline medium in the presence of surfactants. Application of shearing forces, preferably by means of a sand or pearl mill, and maintaining a temperature of 50 °C produces improved pigment quality [187].
3.7.4.3.1 Properties and Application of Pigment Violet 31
Dichloroisoviolanthrone (94) was supplied by the pigment industry in the form of a pigment preparation, which was a speciality product for use in spin dyed viscose fibres. This pigment preparation has been withdrawn from the market. The shade of P.V.31 is a reddish violet, which is distinctly redder than that of the known shade of P.V.23. The pigment exhibits very high quality fastness properties. Depending on the depth of shade, P.V.31 systems equal step 7 to step 8 on the Blue Scale for lightfastness. Their fastness properties in application, especially their fastness to perspiration, dry cleaning and crocking are excellent. P.V.31 shows only poor resistance to vatting. Although the shade of spin dyed articles changes only slightly in an alkaline sodium dithionite solution (60 °C/30 min), colour is clearly transferred onto white spun rayon and cotton.
3.7.4.4 Violanthrone Pigments
Violanthrone, 95, also known as dibenzanthrone, is an isomer of isoviolanthrone (91). In contrast to isoviolanthrone, which has the molecular symmetry C2h and possesses a molecular inversion centre, the violanthrone molecule has C2v symmetry and no molecular inversion centre. Violanthrone is registered as Pigment Blue 65, and just as isoviolanthrone, has an intense blue colour.
Scheme 3.24 Molecular structure of P.B.65.
Violanthrone is traditionally produced by the potash fusion of two molecules of benzanthrone (92) [189]. However, this process yields a crude product, which contains impurities such as isoviolanthrone and 4-hydroxybenzanthrone. A product of better purity is obtained when the condensation of the two benzanthrone molecules is carried out in two steps. In the first step, benzanthrone (92) is dimerized in an alcoholic alkali melt to form 4,4′-bibenzanthronyl (95a), which is then followed by alkaline or acidic ring closure, preferably in the presence of an oxidizing agent [189a, 189b, 189c].

The single-crystal structure determination [189d] confirms that the violanthrone molecule is planar and has C2v symmetry. In the crystal the molecules are stacked on top of each other, as frequently found in pigments. As usual, the molecules are slightly inclined against the stacking direction, see Figure 3.67. The stacks are arranged in a way so that the space is perfectly filled, see Figure 3.68.

Figure 3.67 Crystal structure of violanthrone, P.B.65. Two stacks of molecules are shown.

Figure 3.68 Crystal structure of violanthrone, P.B.65. View along the stacks, showing the perfect filling of space.
P.B.65 serves as a pigment as well as a vat dye and a precursor to other vat dyes. The compound, a violet–black powder, is used as a redish-blue polycyclic colourant. It is resistant against alkali and acids, insoluble in ethanol and exhibits a lightfastness of 8 on the Blue Scale. The pigment is reported to be used for printing inks, colouration of plastics and coatings.
Derivatives of violanthrone with chloro, bromo, nitro or methoxy substituents are produced as vat dyes.
3.8 Dioxazine Pigments
Pigments that are derived from the triphenedioxazine structure (96) have been known since 1928:

The parent compound 96 provides an orange colour, but has no technical importance as a colourant. It was in 1928 that Kränzlein and coworkers (Farbwerke Hoechst) found that sulfonated derivatives of this basic structure provide dyes that can be used as direct dyes on cotton. In the synthesis, sulfonation follows the formation of the heterocyclic ring system.
It was not until 25 years later that a tinctorially strong violet pigment was obtained, by formation and finishing of a water-insoluble 9,10-dichlorotriphenedioxazine derivative [190].
At present, dioxazines no longer enjoy commercial significance as dyes. However, several derivatives are industrially used as violet pigments. The most important is Pigment Violet 23 (see Scheme 3.25).
3.8.1 Preparation of the Starting Materials
One of the primary starting materials for all members of this group is chloranil (tetrachloro-p-benzoquinone). Today, it is customarily prepared by oxidative chlorination of hydroquinone. As an example, a mixture of hydroquinone and concentrated hydrochloric acid is chlorinated initially at 10 °C. After adding water, the reaction mixture is heated and the chlorination continued. Similar but slightly modified routes involve using hydrochloric acid/H2O2 or chlorine/water. Hydroquinone may be replaced by benzoquinone [191].

The synthesis of P.V.23 requires 3-amino-N-ethylcarbazole as starting material. Carbazole is obtained from hard coal tar and is usually ethylated with ethyl bromide or with ethyl chloride. Subsequent nitration and reduction affords 3-amino-N-ethylcarbazole.
3.8.2 Chemistry, Manufacture and Crystal Structures
The commercial dioxazine pigments can be divided into three groups (97–99):

Scheme 3.25 Molecular structure of P.V.23.

Scheme 3.26 Molecular structures of P.Bl.80, P.V.34, P.V.35, P.V.37, and P.V.57.
Two synthetic routes are possible: Route A starts from anilines, route B from o-alkoxyanilines:

The first step, linking the central ring via NH bridges to the terminal aromatic units yielding 2,5-diarylamino-1,4-benzoquinones, typically proceeds in an organic solvent (such as boiling ethanol) below 100 °C and in the presence of an acid trap such as sodium acetate. For P.V.37, chloranil is replaced by 2,5-dichloro-3,6-bis(acetylamino)-1,4-benzoquinone to react with 1,4-diethoxy-2-amino-5-benzoylaminobenzene.
The second step (cyclization) is effected in route A by oxidative condensation under comparatively harsh conditions. The material is treated at 180–260 °C in a high-boiling organic solvent such as chloronaphthalene in the presence of a condensation agent in the form of an acidic catalyst. Suitable agents include benzene sulfochloride, p-tosyl chloride, m-nitrobenzene sulfochloride and aluminium chloride.
In route B cyclization is achieved at comparatively lower temperature, between 170 and 175 °C, and the reaction proceeds at a higher rate than in route A. One of the above-mentioned condensation agents is similarly necessary in this case. o-Dichlorobenzene, for instance, is a suitable organic solvent. The list of solvents for condensation and cyclization also includes trichlorobenzene and nitrobenzene. The reaction conditions are not markedly affected by the type of the substituents R.
In the case of P.V.23 routes A and route B give different isomers [173]:
In earlier times P.V.23 was believed to have the linear structure 97a. In 1987 a single-crystal structure analysis revealed that P.V.23 actually has the S-shaped structure 97 [73]. The linear isomer 97a can be synthesized according to route B. Its structure was also confirmed by single-crystal structure analyses. The linear isomer affords a blue shade and is without any commercial importance.
The procedure for the synthesis of dioxazine pigments in general may be illustrated by the example of Pigment Violet 23. The pigment is synthesized via route A. The technique has not changed much since it was first developed [192]. An excess of chloranil in o-dichlorobenzene is added to 2 mol of 3-amino-N-ethylcarbazole and dry sodium acetate, which acts as an acid trap. The mixture is stirred for 6 h at 60 °C, then within 5 h heated to 115 °C under vacuum, after which benzenesulfochloride is added at the same temperature. Cyclization is achieved by increasing the temperature to 175–180 °C. The reaction mixture is agitated until no more acetic acid appears in the distillation receiver (for 4–8 h). The reaction product is vacuum filtered, residual o-dichlorobenzene removed by steam distillation, and the product washed and dried. A patent that was issued in 1980 claims a much improved yield if the reaction is performed in the presence of only a slight excess of chloranil, provided 0.15–1.8 wt% water is added to the reaction mixture [193].
Production of the crude pigment is followed by finishing. The options include milling the material with salt in a ball mill in the presence of an organic solvent, or treating it similarly in a kneader, or producing a 60–90% sulfuric acid slurry, or using aromatic sulfonic acids.
Replacement of the carbazole groups of P.V.23 by benzimidazolone moieties leads to imidazolone-anellated triphenedioxazine pigments (98), which are also called ‘benzimidazolone-dioxazine’ pigments. As in benzimidazolone hydrazone pigments, the benzimidazolone groups in dioxazine pigments form intermolecular hydrogen bonds, and hence decrease the solubility and enhance the fastness properties. The parent compound (98, R = R′ = H) is so insoluble that it cannot be transformed into a dispersible pigmentary form; thus alkyl groups have to be added to obtain an industrially useful dispersible pigment.
The synthesis is similar to the synthesis of P.V.23. Route A is preferred because the starting materials (substituted benzimidazolones) for route A are more accessible than the corresponding alkoxy-substituted derivatives required for route B. In contrast to P.V.23, route A leads to the linear isomer. The structures of the compounds were confirmed by NMR spectroscopy and crystal structure determinations from X-ray powder data [194].
If the synthesis is started with a mixture of methyl- and ethyl-substituted benzimidazolones, a solid solution of methyl- and ethyl-imidazolone-triphendioxazines is formed, which also contains the mixed methyl/ethyl compound [195]. Interestingly, this solid solution (P.V.57) has a much more reddish shade than the ethyl derivative itself (P.B.80), although the electronic effect of the alkyl substituent is negligible and the crystal structures are isostructural. The colour difference is apparently due to different exciton interactions caused by differences in the positions of neighbouring molecules.
3.8.2.1 Crystal Structures
Pigment Violet 23 forms a criss-cross packing (Figures 3.69 and 3.70) [73]. The molecule is planar; only the ethyl groups protrude from the molecular plane.

Figure 3.69 Crystal structure of P.V.23, determined from single-crystal X-ray data [73].

Figure 3.70 Criss-cross packing in the crystal structure of P.V.23, showing nine molecules.
In Pigment Blue 80 the benzimidazolone moieties of neighbouring molecules are connected via an eight-membered HNCO![]() HNCO ring (Figure 3.71), as is found for many other benzimidazolone compounds. The molecules form chains that arrange in layers (Figure 3.72). As for P.V.23, the molecules are planar, except for the ethyl groups. The packing is quite efficient; the density of P.B.80 is as high as 1.676 g m−3. The above-mentioned solid solution of methyl and ethyl derivatives (P.V.57 (Figure 3.73)) is isostructural to P.B.80 and even has a density of 1.754 g m−3, which is extraordinarily high for an organic compound. The strong intermolecular interactions, the efficient packing and the high density explain the insolubility and the good fastness properties of these compounds [194].
HNCO ring (Figure 3.71), as is found for many other benzimidazolone compounds. The molecules form chains that arrange in layers (Figure 3.72). As for P.V.23, the molecules are planar, except for the ethyl groups. The packing is quite efficient; the density of P.B.80 is as high as 1.676 g m−3. The above-mentioned solid solution of methyl and ethyl derivatives (P.V.57 (Figure 3.73)) is isostructural to P.B.80 and even has a density of 1.754 g m−3, which is extraordinarily high for an organic compound. The strong intermolecular interactions, the efficient packing and the high density explain the insolubility and the good fastness properties of these compounds [194].

Figure 3.71 Crystal structure of P.B.80. One layer of molecules is shown.

Figure 3.72 Crystal structure of P.B.80. View along the layers, showing five molecules.

Figure 3.73 Crystal structure of P.V.57 (solid solution of methyl and ethyl derivatives). One layer of molecules is shown. The methyl and ethyl groups are statistically disordered, but at each location (indicated by the red ellipsis), there is always one methyl group beside one ethyl group.
3.8.3 Properties
Those pigments of the dioxazine series that are of importance to the pigment industry provide clean violet (considerably bluish red) or reddish blue shades. They demonstrate excellent lightfastness and weatherfastness, even in light shades. There is a certain disadvantage due to the fact that most pigments, and especially the most important one, P.V.23, tend to bleed if incorporated into plastics. Imidazolone-anellated dioxazine pigments do not bleed.
Several dioxazine pigments are found in more than one crystal modification. For example, P.B.80 exists in six, P.V.57 in two polymorphic forms [196, 197].
3.8.4 Commercially Available Dioxazine Pigments and Their Application
3.8.4.1 General
At present, this class produces only two representatives that continue to be used industrially: Pigment Violet 23, 51319 and Pigment Violet 37, 51345:


Scheme 3.27 Molecular structures of P.V.23 and P.V.37.
While P.V.23 is used in considerable volume, P.V.37 is a speciality product. Pigments of the P.V.34 and 35 type [198–200] have been discontinued. These pigments, which deviate from P.V.37 in their substitution patterns, are much inferior in terms of tinctorial strength and sometimes also in their migration fastness, especially in plastics.
At the beginning of the twenty-first century, P.B.80 and P.V.57 were introduced into the market [201]. Despite their extraordinary high colour strengths and their excellent fastness properties, their production was suspended a few years later.
3.8.4.2 Pigment Violet 23
P.V.23, also referred to as Carbazole Violet, is a universally useful product. Its colour, a bluish violet shade, is not accessible with other Pigments. P.V.23 is used in almost all media that are typically coloured with pigments. The list of suitable systems ranges from coatings and paints to plastics, printing inks, and other special-purpose media. P.V.23 is entirely fast to many organic solvents. At standardized conditions (Section 1.6.2.1), it is fast to alcohols, esters and aliphatic hydrocarbons as well as to plasticizers such as dibutyl and dioctyl phthalate. Other solvents, such as ketones, are coloured slightly (step 4).
Tinctorially, P.V.23 is an uncommonly strong pigment in almost all media, which qualifies it, even in very small amounts, for use as a shading pigment. Used to a considerable extent as a shading component for paints, P.V.23 adds a reddish tinge to the shade of copper phthalocyanine blue pigments. Although P.V.23 is not quite as lightfast and weatherfast as phthalocyanine blue pigments, it does satisfy most requirements, even very stringent ones. Moreover, P.V.23 is also a useful toning pigment for white enamels. The pigment is particularly important in systems based on TiO2/rutile with their yellowish undertone. Trace amounts of P.V.23 are used to tone the system: only 0.0005–0.05 parts of P.V.23 are required per 100 parts of TiO2. White enamels may also be toned by mixtures of P.V.23 and α-Copper Phthalocyanine Blue. P.V.23 is both completely lightfast and weatherfast, even in very light shades (for instance at 1:3000 reduction with TiO2). Apart from this excellent fastness, P.V.23 has the added advantage of being entirely fast to overcoating. It is therefore used in all types of media throughout the paint industry. The list ranges from air drying house and trade sales paints, that is, decorative (architectural) paints, to general and high grade industrial finishes, such as original automotive (O.E.M) and automotive refinishes. P.V.23 is used in uniformly coloured finishes as well as in metallic finishes. Baking enamels containing P.V.23 are thermally stable up to 160 °C.
Most grades exhibit a high specific surface area, sometimes above 100 m2 g−1. For good dispersion and to avoid flocculation, it is therefore necessary to maintain an adequate pigment/binder ratio. Notably, the required amount of binder is higher in this case than for most other organic pigments.
P.V.23 is a favourite shading pigment for use in emulsion paints, where it lends a reddish tinge to Phthalocyanine Blue shades. Excellent weatherfastness makes it a suitable candidate for exterior application in media based on synthetic resin dispersions. The systems are fast to alkali and plaster.
Plate-out is observed with various hardeners if P.V.23 is incorporated in most types of powder coatings, for instance in epoxy systems. This phenomenon, however, is of no consequence for the applicability of the pigment: only minor amounts of P.V.23 are required in these media to lend a blue shade to white enamels.
The list of suitable applications also includes various special-purpose systems, especially where high lightfastness and durability, but also high heat stability or other perfect fastness properties, are a prime concern. This is true, for instance, for coatings to be applied onto aluminium window blinds. Bilaterally coated, usually in pastel colours, these aluminium strips are baked at 250 °C or less and subsequently subjected to considerable mechanical stress.
P.V.23 is also frequently encountered in plastics. Although plasticized PVC systems containing P.V.23 are not entirely migration resistant, the pigment exhibits unusually high tinctorial strength. Less than 0.3% pigment is needed, for instance, to produce 1/3 SD systems (5% TiO2). The systems demonstrate excellent lightfastness: 1/3 SD specimens equal step 8 on the Blue Scale, while 1/25 SD samples match step 7–8. Moreover, P.V.23 provides excellent weatherfastness and is suited to long-term exposure. It is an important colourant for PVC and PUR plastisols.
In terms of heat stability, 1/3 SD polyolefin systems containing P.V.23 withstand exposure to 280 °C. In lighter tints, this value is appreciably lower: 1/25 SD samples only tolerate up to 200 °C. Dissolution effects shift the shade considerably towards the reddish region, while the pigment maintains its shade at higher temperatures. P.V.23 is an equally tinctorially strong product in these media. 1/3 SD HDPE samples (1% TiO2) are formulated at less than 0.07% pigment. P.V.23 is a typical polycyclic pigment in that it affects the shrinkage of injection-moulded articles made of HDPE and other partially crystalline polymers. This is a tendency that somewhat restricts its use in such media. The pigment concentration in a transparent HDPE system should not be less than 0.05%. P.V.23 systems rapidly lose lightfastness as more white pigment is added. 1/3 SD samples equal step 8 on the Blue Scale, while 1/25 SD specimens only score as high as step 2.
Carbazole Violet is a suitable pigment for transparent polystyrene products. A certain degree of dissolution in this medium at elevated temperatures makes it necessary to limit the use to processes up to approximately 220 °C. P.V.23 is also used in polyester that is processed at very high temperature. The pigment is equally suitable for use in polyester spin dyeing, it is stable to the harsh conditions of the condensation process, during which it is exposed to 240–280 °C for 5–6 h. At low pigment concentrations there is also a colour shift towards redder shades, due to pigment dissolution. P.V.23 is highly lightfast. Although it generally offers high textile fastness properties, the pigment is not entirely fast to dry and wet crocking. P.V.23 also satisfies the thermal and other requirements for use in spin dyed polyacrylonitrile. In this context, the pigment is completely fast to dry and wet crocking. In PP spin dyeing, P.V.23 should not be applied at low concentrations in order to prevent dissolution effects. It provides excellent lightfastness in medium and deep shades. P.V.23 is also recommended for use in spin dyed viscose, where it exhibits good lightfastness and generally excellent fastness properties in application.
Worked into cast resins based on methacrylate or unsaturated polyester, P.V.23 has the advantage of being fast to peroxides, which act as catalysts in these media. The lightfastness of such systems is between step 7 and step 8 on the Blue Scale, both for transparent and opaque colourations.
P.V.23 has stimulated interest throughout the printing ink industry. It is frequently utilized in combination with copper phthalocyanine blue pigments to produce reddish shades of blue. P.V.23 is equally strong in these media. At standardized film thickness, 1/1 SD letterpress proof prints are prepared from inks formulated at only 8.7% pigment, while 1/3 SD specimens require inks containing 5.6%. In terms of lightfastness, these prints equal step 6 to step 7 on the Blue Scale. The prints are very fast to organic solvents and also exhibit excellent application properties, such as fastness to soap, alkali and fat. The prints may be exposed to 220 °C for 10 min or to 200 °C for 30 min and may safely be calendered or sterilized. P.V.23 is also used in textile printing. It is very lightfast and weatherfast in this application and also performs well in terms of other important fastness properties.
Several other media are also pigmented with P.V.23. The list includes office articles and artists' colours, such as drawing inks and fibre-tip pen inks, wax crayons, oil paints, and high quality watercolours, water- or solvent-based pigmented wood stains, cleaning agents and mass coloured paper.
3.8.4.3 Pigment Violet 37
P.V.37 provides a much more reddish shade than P.V.23 but is equally fast to organic solvents. P.V.37, although weaker in some media than P.V.23, is broad in scope. Printing inks based on nitrocellulose are unique in that P.V.37 provides high tinctorial strength and excellent gloss in these media, although the products are somewhat opaque. Despite the fact that, incorporated in oil-based printing inks for purposes such as offset printing, it is somewhat weaker than P.V.23, P.V.37 exhibits high gloss and good flow behaviour. Its main field of application within the printing ink industry is in metal deco printing. Its fastness properties in application equal those of P.V.23.
In paints, P.V.37 also exhibits a considerably more reddish shade and reduced tinctorial strength. Similar observations apply to its colouristic properties in polymers. In plasticized PVC, P.V.37 has the advantage of being considerably more migration resistant than P.V.23; in fact, it is almost entirely fast to migration. Since P.V.37, like P.V.23, dissolves to some extent in polymers, it loses much of its heat stability as the white pigment concentration in a system is increased. Similar observations apply to polyolefin systems: the shade provided by P.V.37 is considerably more reddish and the tinctorial strength much inferior to that of P.V.23: 0.09% P.V.37 is required to afford 1/3 SD in HDPE systems (1% TiO2). Such systems are heat stable up to 290 °C but, notably, the heat stability declines rapidly in certain concentration ranges, a tendency that is also observed with P.V.23. Like P.V.23, P.V.37 also considerably affects the shrinkage of HDPE and other partially crystalline injection-moulded plastics. P.V.37 is also recommended for use in PP spin dyeing. As with P.V.23, systems formulated at low concentrations tend to undergo a colour shift due to pigment dissolution.
3.8.4.4 Pigment Violet 57
P.V.57 exhibits a bright reddish violet shade, and a high colour strength. The fastness properties in paints are as high as for P.B.80, which makes the pigment suitable for automotive coatings and paints.
3.8.4.5 Pigment Blue 80
P.B.80 is a reddish blue pigment, shadewise between P.V.23 and P.B.60 (Indanthrene Blue) [201]. The pigment shows outstanding colour strength, which even surpasses P.V.23 in this respect. In PVC the colour strength exceeds that of P.V.23 by about 30%. In paints, the compound can be mixed with 30 parts of TiO2 to get 1/3 standard depth, and with 300 parts of TiO2 to get a shade of 1/25 standard depth. Apparently P.B.80 has the highest colour strength of all commercial organic pigments. Lightfastness and weatherfastness are excellent, both in solid and metallic applications. The light fastness is at the highest grade (grade 8), even in 1/25 standard depth. The pigment passes the three-year Florida exposure test even better than P.B.60. The high transparency makes the pigment suitable for all kinds of effect shade applications. Because of its weather fastness, P.B.80 can even be used for shading silver-metallic coatings.
The pigment is completely resistant to the major solvents used in paint applications and the fastness to overpainting is excellent even at baking temperatures of 180 °C. The heat stability is outstanding and, thus, P.B.80 can be used in coil coating applications without any restriction. In principle, the heat stability for powder coating applications is also excellent, but the surface treatment of the pigment may cause some incompatibility in certain powder coating systems. P.B.80 is recommended for all kinds of high-performance applications, such as OEM, refinish and coil coating.
3.9 Quinophthalone Pigments
3.9.1 Chemistry, Manufacture and Crystal Structures
In 1882 E. Jacobson fused phthalic anhydride with quinoline bases obtained from coal tar, which also contained quinaldine (100). He thus obtained quinophthalone (101). Quinophthalone derivatives bearing sulfonic or carboxylic acid functions represent suitable anionic dyes. Derivatives carrying basic side chains containing quaternary nitrogen, on the other hand, provide cationic dyes. The compounds are used especially as disperse dyes [202].
Compound 101, the parent structure to all pigments described in this section, continues to be prepared by fusion or, even better, by treating quinaldine with phthalic anhydride in an inert high-boiling solvent at 200–220 °C:

The structure was elucidated by Eibner between 1904 and 1906. The formation of the intramolecular hydrogen bond was proven through IR and nuclear magnetic resonance spectroscopy (NMR) [203]. In the solid state, the compound exists in the keto form 101b.
Quinophthalone molecules are too soluble in various media to be used under normal application conditions. Both solvent and migration resistance may be enhanced by enlarging the molecule; the options are as follows:
- introducing suitable substituents,
- condensing quinaldines with pyromellitic dianhydride instead of phthalic anhydride,
- doubling the molecule via diamide bridges.
These methods produce yellow to red compounds that exhibit satisfactory pigment properties.
The list of suitable substituents includes acylamino groups, but especially halogen atoms such as chlorine or bromine. Halogenated derivatives are obtained from tetrahalogen phthalic anhydride or naphthalene-2,3-dicarboxylic anhydride, by reaction with quinaldine (or one of its derivatives). A patent has been issued describing the reaction of 8-aminoquinaldine with twice the stoichiometric amount of tetrahalogen phthalic anhydride in an inert solvent in the presence of zinc chloride [204]. This route makes it possible to introduce eight halogen atoms into the molecule (102), resulting in greenish yellow pigments:

Scheme 3.28 Synthesis of P.Y.138.
Pigment Yellow 138, C.I. 56300, has the chemical structure 102 with X = Cl [205].
The synthesis of halogenated compounds (102), for instance P.Y.138, can also be achieved by stepwise heating of tetrachlorophthalic anhydride and 8-aminochinaldine in molten benzoic acid from 125 to 140 and then to 160 °C [207].
Another publication describes the conversion of 8-chloro-5-aminoquinaldine with possibly halogenated phthalic anhydride [208]. These publications also discuss the fact that halogenation of the phthalimide ring improves the lightfastness of the product, but on the other hand that lightfastness is diminished if substitution is made to the benzene ring of the quinaldine system. Apart from compounds bearing an OH group in 3′-position, 4′-OH and 4′-acylamino derivatives also show enhanced lightfastness. Condensation of 3-hydroxyquinaldine or its derivatives with pyromellitic dianhydride affords bis-quinophthalones (103). These are very lightfast pigments affording yellow, red or brown shades:

The quinophthalone structure may also be doubled through condensation of two equivalents of 3-hydroxy-quinophthalone carboxylic acid chloride with aromatic diamines [209]. The reaction affords orange pigments (104). The acid chloride is prepared from 3-hydroxyquinaldine-4-carboxylic acid and benzene-1,2,4-tricarboxylic acid:

The crystal structure and the tautomeric state of P.Y.138 were determined in 2015 by a combination of X-ray powder diffraction, solid-state NMR and dispersion-corrected density functional calculations (DFT-D) [209a]. Before these investigations, three different tautomers could be found in the literature:

The solid-state NMR analysis and the DFT-D calculations revealed that P.Y.138 exists in the NH-tautomeric form in the solid state. The proton of the NH group forms an intramolecular hydrogen bond with the CO group of the indanedione fragment (Figure 3.74). The central quinolene system is coplanar with the indanedione system (right), but the phthalimide group (left) is strongly rotated out of the plane, with a dihedral angle of 57°.

Figure 3.74 P.Y.138. Molecular structure in the solid state [209a].
The molecules are arranged in sheets, which are connected by chlorine-chlorine contacts (Figure 3.75).

Figure 3.75 Crystal structure of P.Y.138. The molecules in the front are drawn with filled circles, those one in the back with open circles. Note that the molecules are not planar, but twisted (see Figure 3.74).
3.9.2 Properties and Application
Quinophthalone pigments are marketed in very limited number. The pigments provide yellow to red shades and are used primarily to colour paints and plastics.
3.9.3 Commercially Available Quinophthalone Pigments
3.9.3.1 Pigment Yellow 138
P.Y.138 (Scheme 3.28) affords exceedingly lightfast and weatherfast greenish yellow shades with good heat stability. Its main fields of application are in paints and in plastics.
Most commercial P.Y.138 types have low specific surface areas of approximately 25 m2 g−1 and consequently provide good hiding power, a feature that qualifies them for use in opaque systems. In this field, P.Y.138 competes with opaque yellow pigments of other classes, which, although tinctorially weaker, sometimes offer better hiding power and cleaner shades. Paints formulated at higher pigment concentrations show good flow behaviour. P.Y.138 is entirely or at least largely resistant to a large number of organic solvents, such as alcohols, esters, ketones and aliphatic as well as aromatic hydrocarbons. Oven drying systems containing P.Y.138 are fast to overcoating at common baking temperatures; the pigment is thermostable up to 200 °C.
Full shades and similar deep shades exhibit excellent weatherfastness, but rapidly decrease in tints made by adding TiO2. The cleanness of full shades increases noticeably by exposure to weather. There are certain binders in which P.Y.138 tends to chalk if employed at high concentration.
P.Y.138 is primarily applied in industrial finishes for commercial vehicles, automotive refinishes and original automotive finishes. Incorporated in these systems, P.Y.138 is acid and alkali fast. However, exterior house paints that are applied onto a basic substrate show some sensitivity to alkali.
Types that feature slightly higher specific surface areas are more transparent and demonstrate somewhat different colouristic properties. Such types are not only tinctorially stronger and more greenish but also somewhat less fast to light and weather than opaque varieties, and their flow behaviour is less favourable.
Another main field of application for P.Y.138 is in plastics, in which it demonstrates average to good tinctorial strength. 1/3 HDPE samples (1% TiO2) are formulated at approx. 0.2% pigment. Such systems are heat stable up to 290 °C, as are full shades. 1/25 SD specimens only withstand 250 °C. The pigment nucleates in this polymer, as in other partially crystalline plastics, that is, it affects the shrinkage of injection-moulded articles. This effect becomes markedly less noticeable as the processing temperature rises. In terms of lightfastness, full shades equal step 7–8 on the Blue Scale, while white reductions down to 1/25 SD match step 6–7. P.Y.138 is an equally suitable colourant for polystyrene, ABS and various other plastics, such as polyurethane foam. A wide variety of pigment preparations are supplied for various areas of application within the paint and plastics industry. These preparations have the advantage of containing already dispersed pigment.
3.10 Isoindolinone and Isoindoline Pigments
3.10.1 General
The pigments described in this section are azomethine and methine pigments, which in the classification system adopted in this book are formally located between hydrazone pigments and polycyclic pigments.
The common structural feature underlying these pigments is the isoindoline ring (X1 = X3 = H):

Positions 1 and 3 are substituted further.
Depending on the atom X, two classes are defined:
- azomethine derivatives (X1 = O or N-; X3 = N-), called isoindolinone pigments;
- methine derivatives (X1 = X3 = C<), called isoindoline pigments.
3.10.1.1 Isoindolinone Pigments
The important group of tetrachloroisoindolinone pigments are azomethine derivatives. They were introduced in the mid-1960s.
Formally, these pigments are disazomethine types, obtained by condensation of primary aromatic diamines with two equivalents of tetrachloroisoindoline-1-one:

In fact, two tetrachlorophthalimide molecules are linked via a disazomethine bridge:

Patents concerning isoindolinone type dyes and pigments were first published by ICI in 1946. Routes to other such pigments followed in 1952 and 1953 (see Reference [210]). All of these routes started either from the unsubstituted phthalimide or from a phthalimide backbone carrying only one or two substituents (105):

Only very few of these yellow and orange pigments have stimulated interest in terms of technical application, for example P.Y.173 (106) [211]:

Scheme 3.29 Molecular structure of P.Y.173.
This is a yellow pigment, obtained by condensing 2,5-dichloro-1,4-diaminobenzene dihydrochloride with 3-iminoisoindolinone in chlorobenzene at 140 °C:

A decisive step towards the development of isoindolinone pigments is credited to Geigy. Patents published in the late 1950s describe pigments with the basic structure 105, using tetrachlorophthalimide as starting material.
The technique of ‘perchlorinating’ the two aromatic rings surprisingly enhances both the tinctorial strength and the fastness properties of the product considerably. This quality increase is notably attributed to factors that are described in detail in Section 3.10.2.
3.10.1.2 Isoindoline Pigments
The methine type consists of isoindoline derivatives. One or usually two equivalents of a compound containing an activated methylene moiety are attached to one equivalent of isoindoline. The list of compounds featuring activated methylene groups includes cyanacetamide or heterocycles such as barbituric acid or tetrahydroquinolinedione.
Notably, none of these pigments feature chlorine substitution on the aromatic system.
3.10.2 Chemistry, Manufacture and Crystal Structures
3.10.2.1 Isoindolinone Pigments
Commercially used tetrachloroisoindolinone pigments have the general chemical structure 107 [210]:

The synthetic route generally involves condensing two equivalents of 4,5,6,7-tetrachloroisoindoline-1-one derivatives with one equivalent of an aromatic diamine in an organic solvent. Suitable tetrachloroisoindoline-1-one derivatives are substituted in 3-position, which is occupied either by two monovalent groups (A) or one divalent group (B). A may represent a chloro atom or a CH3O group, while B usually represents a NH moiety:

Primary starting materials for this condensation reaction are 3,3,4,5,6,7-hexachloroisoindoline-1-one (108) and 2-cyano-3,4,5,6-tetrachlorobenzoic acid methyl ester (109):

Compound 108 directly reacts with a diamine to afford the desired pigment. Two pathways have been found effective for the synthesis of 108. Tetrachlorophthalimide may be chlorinated either:
- with one equivalent of phosphorus pentachloride in phosphorus oxychloride;
- with two equivalents of phosphorus pentachloride, affording 1,3,3,4,5,6,7-heptachloroisoindolenine (110) as an intermediate, which is to be converted into 108 with one equivalent of water or alcohol:

Compound 109 reacts with ammonia or alkali alcoholate to form the corresponding activated compounds 111 and 112:

Compound 109 is synthesized from 3,3,4,5,6,7-hexachlorophthalic anhydride, which is treated with ammonia to yield the ammonium salt of 2-cyanotetrachlorobenzoic acid. After being converted into the sodium salt, the thus-prepared intermediate is reacted with methyl halogenides or dimethyl sulfate to form the methyl ester 109.
The synthetic route may be exemplified by the preparation of a tetrachloroisoindolinone pigment. A mixture of one mole of 1,4-diaminobenzene in o-dichlorobenzene with a solution of two moles of 3,3,4,5,6,7-hexachloroisoindoline-1-one in o-dichlorobenzene is heated to 160–170 °C for 3 h. Closed filtration equipment is used to filter the hot product, which is then washed with o-dichlorobenzene and alcohol, dried and milled. The resulting product is a reddish yellow pigment with the structure 113, which is registered as P.Y.110:

Scheme 3.30 Molecular structure of P.Y.110.
o-Dichlorobenzene can be substituted advantageously by 1,2,3-trimethylbenzene [212].
The following published examples illustrate some developments in the field of tetrachloroisoindolinone pigments.
In 1973, a patent describing a new synthetic method was issued to Dainippon Ink [213]. According to this publication, the bisacylated compound 114 is prepared from easily accessible tetrachlorophthalic anhydride by reacting with an aromatic diamine at a 2 : 1 molecular ratio in the presence of ammonia and phosphorus pentachloride. Additional phosphorus pentachloride in a high-boiling solvent, such as trichlorobenzene, effects ring closure of 114 to form the tetrachloroisoindolinone pigment:

By finishing, different polymorphic forms can be obtained. A β-modification of P.Y.110 is formed by heating the known α-modification to temperatures between 200 and 350 °C in one of various high-boiling solvents [214]. A γ-modification of P.Y.110 is apparently accessible by slowly hydrolysing the corresponding potassium salt, which is obtained by the usual methods, with water [215].
Other publications describe mono and disazomethine pigments featuring heterocyclic ring systems [216]. There are also patents concerning bisiminoisoindolenine pigments [217].
There is a large number of studies concerning the synthesis of new chemical species based on the isoindolinone system. The most notable publications are those that refer to modifications of the bridging diamine between the two isoindolinone systems. The following diamine derivatives have been mentioned:

[218–221]
Moreover, there are patents claiming monohydrazone and dihydrazone pigments, starting from compounds such as

acting as diazo components [222], or, after appropriate acetoacetylation with diketene, as coupling components [223].
The crystal structure of the commercial α-phase of P.Y.110 has been determined from X-ray powder data by a combination of molecular quantum-mechanical calculations, lattice-energy calculations and intuition [224]. The central phenylene group is rotated out of the plane formed by the isoindolinone groups by 52°. The molecules are arranged in chains connected by double hydrogen bonds, see Figures 3.76 and 3.77.

Figure 3.76 Crystal structure of the α-phase of P.Y.110 [224, 224a]. Two chains are shown.

Figure 3.77 Crystal structure of the α-phase of P.Y.110 [224]. View along the chains.
Treatment of the α-phase of P.Y.110 with solvents at elevated temperatures leads to the formation of the β-phase. The crystal structure of the β-phase was also determined by X-ray powder diffraction [224a]. The angle between the isoindolinone groups and the central phenylene unit increased to 62°. The molecules are arranged in chains similar to the α-phase, but the mutual arrangement of the chains is different (Figure 3.78).

Figure 3.78 Crystal structure of the β-phase of P.Y.110 [224a]. View along the chains.
3.10.2.2 Isoindoline Pigments
Several publications describe various isoindoline pigments with different chemical structures. Such species consist of an isoindoline ring attached to two methine bridges.
These pigments have the following general chemical structure:

R1 through R4 represent CN, CONH-alkyl, or CONH-aryl. R1 and R2 on the one hand and R3 and R4 on the other hand can be also members of a heterocyclic ring system.
Examples, such as 115 type pigments, have been described by BASF and Ciba-Geigy [225, 226]:

Scheme 3.31 Molecular structures of P.Y.185 and P.O.66.
in which R5 stands especially for a methyl group or possibly for a substituted phenyl moiety, or for the almost symmetrically substituted compound 116 (P.O.66) [227]. Pigments with this structure afford yellow shades.
Likewise, suitable starting materials are iminoisoindolines, especially diiminoisoindoline (1-amino-3-iminoisoindolenine), which reacts with a cyanoacetamide NCCH2CONHR5 to afford a mono-condensation product 117. Further reaction with a compound containing an activated methylene group (such as cyanoacetamide derivatives, barbituric acid) yields the desired pigment 115:
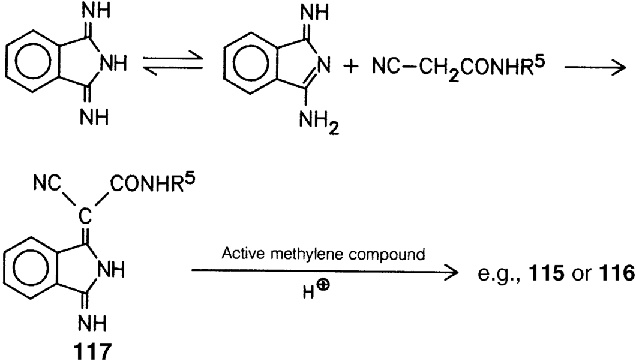
It is also possible to perform a simple one-step synthesis by adjusting the pH (initially pH 8–11, then pH 1–3).
The manufacture of P.Y.139 may serve as an example for the synthesis of a methine pigment. P.Y.139 is the condensation product of diiminoisoindoline with 2 mol of barbituric acid [228]. Gaseous ammonia is introduced into o-phthalodinitrile suspended in ethylene glycol, forming diiminoisoindoline. This compound is transferred into a mixture of an aqueous solution of barbituric acid, formic acid and a surfactant. After boiling at reflux for 4 h, the hot reaction mixture is filtered, washed to neutrality and free of auxiliaries, and dried. The resulting pigment is a greenish yellow compound, P.Y.139:

Scheme 3.32 Synthesis of P.Y.139.
It is also possible to synthesize P.Y.139 from one equivalent of 1-amino-3-cyaniminoisoindolenine:

and two equivalents of barbituric acid in aqueous solution [229].
In another process [230] phthalonitrile, dissolved in an alcohol, is reacted with a base and, without isolating, condensed with barbituric acid in a mixture of formic acid and water.
A one-step synthetic route to methine pigments, starting from o-phthalonitrile, has been described by BASF [231].
An asymmetric isoindoline pigment preparation has been claimed, starting from 1,2-dicyanobenzene, which is reacted in alcohol and a base, followed by addition of a cyanomethylene compound and subsequently a barbituric acid derivative, whereby all reactions are accomplished without isolation of the corresponding intermediates [232].
Ciba-Geigy [233] has investigated other methine pigments, including bismethine compounds with one or two non-halogenated isoindolinone ring systems.
The crystal structure of P.Y.139 was determined from X-ray powder diffraction data by P. Erk [224, 224a, 234] using a combined refinement against the X-ray powder data and the lattice energy. The compound crystallizes in the rare space group Cmce with eight molecules per unit cell. The molecules are situated on a mirror plane going vertically through the isoindoline moiety. The molecule is not planar. Because of steric hindrance between the isoindoline and the neighbouring CO groups, the barbituric acid groups are rotated out of the isoindoline plane by 22° (Figure 3.79). A strong two-dimensional hydrogen bond network connects the molecules to their neighbours (Figure 3.80), which explains the good fastness properties of this comparatively small molecule. The network is not planar, but slightly wavy.

Figure 3.79 Molecular structure of P.Y.139. The barbituric acid moieties are tilted out of the plane of the isoindoline moiety. The isoindoline fragment is drawn in green.

Figure 3.80 (a) Hydrogen-bond network in P.Y.139 (only one layer drawn; the next layer is rotated by 180°); (b) side view onto the layers.
The crystal structures of P.Y.185 and P.R.260 were determined from single-crystal data [234]. In P.Y.185 the molecules are entirely planar. Packing effects obviously induce the concurrence of the plane of the barbituric acid group and the isoindoline plane. The hydrogen bond pattern of the barbituric acid fragments is the same as in P.Y.139. On the cyanomethylene side of the molecule a single hydrogen bond is formed between the NH group and the CN group of a neighbouring molecule. The molecules are arranged in layers, which are (in contrast to P.Y.139) completely planar.
In P.R.260 (118) the barbituric acid moiety is twisted against the isoindoline moiety, as in P.Y.139. In contrast, the cyanomethylenequinazolone fragment, which is sterically less demanding, is coplanar with the isoindoline. The quinazolone group is connected to the barbituric acid group of a neighbouring molecule via double hydrogen bonds, which form an eight-membered ring as for example in P.Y.139, P.Y.185 and P.Y.213. The barbituric acid group forms two of these hydrogen bonded rings, one with the quinazolone fragment, and the second one with a barbituric acid group of another neighbouring molecule. Thus the molecules form non-planar layers.

Scheme 3.33 Molecular structure of P.R.260.
3.10.3 Properties
3.10.3.1 Isoindolinone Pigments
Tetrachloroisoindolinone pigments are available in shades from yellow to orange to red and brown. Commercially important pigments cover the spectral range from greenish to reddish yellow. Interestingly, the corresponding non-chlorinated compounds produce yellow shades, which undergo a bathochromic shift as chlorine atoms are introduced. In terms of explanation, two arguments should be considered. Steric problems, on the one hand, prevent the pigment molecules from assuming a coplanar arrangement but, on the other hand, absorption in the visible portion of the spectrum is possible only in the presence of electron conjugation. It is therefore reasonable to assume that the typical pigment molecule represents a donor–acceptor complex, with the amine functioning as a donor and the tetrachlorinated nucleus acting as an acceptor. The central part, that is the diamine moiety, contributes to the colour insofar as higher conjugation and a large number of π-electrons cause a noticeable bathochromic shift in diamines.
The pigments are only sparingly soluble in most solvents, which makes for good migration fastness. Stability to acids, bases and oxidizers, as well as to reducing agents is very good. The pigments exhibit good heat stability and melt around 400 °C. They are also excellently fast to light and weather, especially in white reductions.
3.10.3.2 Isoindoline Pigments
Isoindoline pigments afford yellow, orange, red and brown shades. They match the above-mentioned tetrachloroisoindolinone pigments in terms of solvent and migration fastness, heat stability and chemical inertness. Likewise, isoindoline pigments demonstrate good lightfastness and weatherfastness. Several azomethine pigments have also been found to be polymorphous.
3.10.4 Application
Isoindolinone and isoindoline pigments are high quality products. They are used in general and high grade industrial paints including original automobile and automotive refinishes, in plastics and for spin dyeing, and in high grade printing inks, especially for metal deco, laminated plastic sheets, and in printing inks for bank notes and securities.
3.10.5 Commercially Available Isoindolinone and Isoindoline Pigments
3.10.5.1 General
Heading the list of commercially available pigments are yellow representatives, such as Pigment Yellow 109, 110, 139, 173 and 185, as well as Pigment Orange 61, 66 and 69, and Pigment Red 260. Another available type is Pigment Brown 38. Pigments providing other shades have failed to gain much commercial impact.
Table 3.9 lists examples of tetrachloroisoindolinone and isoindoline pigments (for general literature, see also Reference [235]). P.Y.173 is also available as a mixture of 30% with 70% of [236]:

Table 3.9 Examples of tetrachloroisoindolinone and isoindoline pigments.
| C.I. Name | C.I. Constitution Number | Structure | Shade | Reference |
| a) Examples of the azo methine type | ||||
| P.Y.109 | 56284 | 
|
greenish yellow | [237, 238] |
| P.Y.110 | 56280 | 
|
reddish yellow | [237, 238] |
| P.Y.173a) | 561600 | 
|
greenish yellow | [235] |
| P.O.61 | 11265 | 
|
orange | |
| – | – | 
|
red | [235] |
| b) Examples of the methine type | ||||
| P.Y.139 | 56298 | 
|
reddish yellow | [238, 239, 240] |
| P.Y.185 | 56280 | 
|
greenish yellow | |
| P.O.66 | 48210 | 
|
yellowish orange | [227] |
| P.O.69 | 56292 | 
|
yellowish orange | [241] |
| P.R.260 | 56295 | 
|
yellowish red | |
| P.Br.38 | 561660 | 
|
yellowish brown | [242,243] |
| a) Also available as mixture (30%) with 70% of the monochloro compound. b) In the original literature [242] no information is given, which of the three possible isomers shown below is formed in the syntheses. A later publication written by scientists from the manufacturer (BASF) [243] shows the E,E-isomer. 
|
||||
3.10.5.2 Pigment Yellow 109
P.Y.109 affords clean, greenish shades of yellow. It is recommended for use in paints, plastics and printing inks. The paint industry uses P.Y.109 primarily to colour various high grade industrial finishes. The list includes, for instance, original automotive and automotive refinishes. P.Y.109 imparts very deep colours on automobile (OEM) finishes, particularly in combination with inorganic pigments. Green shades are produced in conjunction with blue compounds, such as phthalocyanine pigments. Full shades and related shades, reduced up to 1 : 1 with TiO2, darken upon exposure to light. Depending on the type, the lightfastness in full shades (5%) equals step 6–7 to step 7 on the Blue Scale. Samples that are reduced with TiO2 at a ratio of 1 : 3 to 1 : 25 match step 7–8. Both lightfastness and weatherfastness deteriorate rapidly with further reduction. P.Y.109 is fast to overcoating, its heat stability satisfies almost all requirements. Incorporated in an alkyd-melamine system, for instance, 1 : 50 reduction ratios with TiO2 still afford products that are stable up to almost 200 °C. P.Y.109 is used not only in industrial finishes but also in architectural and emulsion paints.
Its main field of application within the plastics industry is in polyolefin colouration. 1/3 SD samples containing 1% TiO2 withstand up to 300 °C, while 1/25 SD specimens are heat stable up to 250 °C. The shade, however, is less clean at this temperature, it becomes duller. In injection moulding, P.Y.109 considerably affects the shrinkage of the plastic. It is a tinctorially weak pigment: 0.4% pigment is required to produce 1/3 SD HDPE samples containing 1% TiO2.
P.Y.109 is fast to migration in plasticized PVC and shows good heat stability. In white reductions (1/25 SD), it is fast up to 160 °C. A noticeable colour change is observed after a 10 min exposure to 180 °C, while full and related shades change colour only at 200 °C. In terms of lightfastness, P.Y.109 equals step 6–7 on the Blue Scale in white reductions (1/3 to 1/25 SD with 5% TiO2). This is slightly inferior to the lightfastness of the redder dihydrazone condensation pigments P.Y.128, 94 and 93. P.Y.109 exhibits good weatherfastness in rigid PVC, but does not satisfy the requirements of long-term exposure. This is especially true for white reductions with a higher TiO2 content. Moreover, P.Y.109 is used to colour polystyrene and other plastics, to rubber, polyurethane, aminoplastic resins and unsaturated polyesters. It is also recommended for use in polypropylene spin dyeing.
P.Y.109 colours high grade products throughout the printing ink industry, but it is in direct competition with a large number of similarly coloured products of various classes. Prints containing P.Y.109 exhibit good fastness to several organic solvents. They are not, however, entirely fast to the DIN 16 524/1 standard solvent mixture. The prints show good heat stability, are fast to overcoating and may safely be sterilized. This makes P.Y.109 a suitable candidate also for metal deco printing. Its lightfastness is good.
3.10.5.3 Pigment Yellow 110
P.Y.110 affords very reddish shades of yellow. Good fastness properties make it a widely used pigment.
The paint industry uses the relatively weak P.Y.110 frequently as a colourant for industrial finishes, especially for high grade finishes. The pigment is very lightfast and weatherfast, which also makes it a suitable product for automotive finishes, for instance original automotive finishes. High transparency is an asset in metallic shades. Deep colours, for instance at 1 : 1 TiO2 reduction, initially brighten as they are exposed to light and weather, but further exposure only has a minor effect on the intensity of the colour. P.Y.110 turns redder as it is combined with TiO2. It is completely fast to overcoating and heat stable up to 200 °C for 30 min. The pigment is also applied in emulsion paints and in architectural paints.
Incorporated in plastics, P.Y.110 is highly heat stable and possesses excellent lightfastness and weatherfastness. Full shades and related shades up to 1/3 SD in plasticized PVC withstand exposure to 200 °C for 30 min. Colourations up to 1/25 SD containing 5% TiO2 equal step 7–8 on the Blue Scale for lightfastness. P.Y.110 also shows excellent durability in rigid PVC and impact resistant PVC types, as well as in plastisols for coil coating. It satisfies the requirements for long-term exposure. P.Y.110 is one of the most lightfast and weatherfast organic yellow pigments known. It shows average tinctorial strength. Between 1.4 and 1.9% pigment is needed to formulate 1/3 SD colourations with 5% TiO2, depending on the type. Comparative values are listed for several other pigments covering the same range of shades.
P.Y.110 is fast to bleeding. Its high heat stability is used to advantage in polyolefins. 1/3 SD HDPE samples maintain their colour up to 290 °C, while 1/25 SD specimens tolerate up to 270 °C for 5 min. The colour turns duller, indicating pigment decomposition. In HDPE melt extrusion, P.Y.110 has considerable effect on the shrinkage of this partially crystalline polymer at processing temperatures between 220 and 280 °C. The pigment is also very lightfast in polyolefins.
P.Y.110 lends colour to polystyrene and styrene containing plastics. It is a suitable candidate for unsaturated polyester and other cast resins, as well as for polyurethane. P.Y.110 is used to an appreciable extent in polypropylene spin dyeing, it is very lightfast in this medium. It is utilized in polyacrylonitrile spin dyeing and sometimes also in polyamide. Its fastness properties, however, especially its lightfastness, do not meet special application conditions (Section 1.8.3.8).
The printing ink industry applies P.Y.110 in all types of printing, provided the pigment satisfies the demands. The prints are resistant to many organic solvents, including the DIN 165224/1 standardized solvent mixture. Prints made from P.Y.110 are fast to clear lacquer coatings, sterilization and are very heat stable. 1/1 to 1/25 SD letterpress proof prints equal step 7 on the Blue Scale for lightfastness.
Likewise, P.Y.110 is found in various other media, such as solvent-based wood stains.
3.10.5.4 Pigment Yellow 139
P.Y.139 is a reddish yellow pigment, used in plastics, paints, and printing inks. The commercial types exhibit a wide variety of particle size distributions and accordingly demonstrate very different colouristic properties, which is especially true for the hiding power. The opaque version is considerably redder. Incorporated in a paint, it is less viscous, which makes it possible to increase the pigment concentration without affecting the gloss of the product.
P.Y.139 is sometimes used in conjunction with inorganic pigments for paints, especially to replace Chrome Yellow pigments. The systems are fast to overpainting (up to 160 °C for 30 min), but they are not entirely fast to acids. The pigment performs very poorly in contact with alkali, therefore it is not suitable for use in amine hardening systems or in emulsion paints that are to be applied on alkaline substrates.
The paint industry uses P.Y.139 to colour general and high grade industrial finishes, including automotive finishes, up to a 1 : 5 TiO2 reduction ratio. The pigment is very lightfast and weatherfast, but its full shade and related shades darken noticeably upon exposure to light and weather. The opaque version performs better than the transparent one. The difference between the two, depending on the depth of shade, is 1/2 to 1 step on the Blue Scale. Incorporated in an alkyd-melamine resin, for instance, 1/3 SD samples of the opaque type equal step 7–8 on the Blue Scale, while corresponding transparent specimens match step 6–7.
P.Y.139 shows average tinctorial strength in plastics. Approximately 1% pigment is required to produce 1/3 SD samples in plasticized PVC containing 5% TiO2. 1/3 SD HDPE specimens containing 1% TiO2 are made from 0.2% pigment. Comparative values are listed for various other pigments. P.Y.139 is bleed resistant in plasticized PVC. It is often used in combination with inorganic and organic pigments, both in plasticized PVC and in PE. HDPE systems containing P.Y.139 in its transparent form (0.1%) and in white reductions (0.1% Pigment + 0.5% TiO2) equal step 7–8 on the Blue Scale for lightfastness. Full shades darken considerably. P.Y.139 is not durable enough to satisfy more stringent requirements. 1/3 SD HDPE samples withstand exposure to 250 °C, while 1/25 SD specimens are heat stable up to 260 °C. The colour becomes duller at higher temperatures, which is a result of pigment decomposition. P.Y.139 is an equally suitable candidate for PP spin dyeing. It is also found in PUR and in unsaturated polyester.
The printing ink industry utilizes P.Y.139 to colour high grade printing products. 1/3 SD letterpress proof prints equal step 7 on the Blue Scale for lightfastness.
3.10.5.5 Pigment Yellow 173
P.Y.173, an isoindolinone pigment, affords somewhat dull, greenish yellow shades. It shows average fastness to organic solvents, especially to alcohols (ethanol), esters (ethyl acetate) and ketones (including methyl ethyl ketone and cyclohexanone). Its solvent resistance equals step 3 on the 5 step scale. P.Y.173 is almost completely fast to mineral spirits and xylene.
P.Y.173 is weatherfast enough to be recommended for high grade industrial finishes, including automotive finishes, and for architectural paints. Notably, P.Y.173 is used advantageously to produce metallic effects in coatings. It is specifically recommended as a shading pigment, sometimes in conjunction with transparent iron oxide, to adjust flops of hue (Section 3.1.5.3). It is a colourant with high tinctorial strength compared to other pigments covering the same range of shades.
Alkyd-melamine resin systems may safely be overcoated; they withstand exposure up to 160 °C for 30 min. 1/3 SD specimens are heat stable up to a maximum of 180 °C. P.Y.173 shows excellent lightfastness and weatherfastness in white reductions, even as larger amounts of TiO2 are added.
The plastics industry uses P.Y.173 to colour various polymers. Incorporated in plasticized PVC, P.Y.173 is one of the tinctorially weaker pigments of its class: 1.8% pigment is required to produce 1/3 SD specimens containing 5% TiO2. The systems are almost completely fast to overcoating. P.Y.173 demonstrates excellent lightfastness and durability in rigid PVC and in PVC plastisols intended for coil coating.
Tinctorially, P.Y.173 is equally weak in PE. It exhibits good heat stability. 1/25 SD samples reduced with TiO2 retain their colour up to 300 °C. 1/3 SD specimens containing 1% TiO2 increase in strength as the temperature exceeds approximately 260 °C. At 300 °C, for instance, as more of the pigment dissolves, such colourations not only exhibit higher cleanness but are also approximately 35% stronger than at 260 °C.
In printing inks P.Y.173 it is a very green shade opaque yellow with excellent light, weather and solvent fastness properties, but low colour strength. It is recommended for paste and liquid inks where high fastness properties are required as well as for water based decorative inks.
3.10.5.6 Pigment Yellow 185
P.Y.185, a comparatively recent product, is an isoindoline pigment and provides clean greenish shades of yellow. Its main field of application is in packaging printing inks. P.Y.185 confers good tinctorial strength and high gloss on prints made from NC-based inks. Its tinctorial strength in print exceeds that of P.Y.17, a pigment of the diarylide yellow pigment series, which provides the same hue. In terms of tinctorial strength, P.Y.185 approaches the equally strong but redder P.Y.74. Likewise, the pigment exhibits high tinctorial strength in offset application. Prints made from P.Y.185 perform like those made from monohydrazone yellow pigments in terms of resistance to organic solvents, including fastness to the standardized DIN 16 524 solvent mixture. The same is true for the fastness to packaged goods, such as butter. However, P.Y.185 performs unsatisfactorily towards alkali (step 2). The prints are heat stable and show good lightfastness. 1/3 SD letterpress proof prints, for instance, equal step 5–6 on the Blue Scale for lightfastness. The commercial type provides good transparency.
Recently, special types for the paint industry and for application media apart from the areas of printing inks, paints and plastics were released to the market. The paint grade is tinctorially very strong. Its tinctorial strength in an alkyd melamine resin baking enamel is more than double as high as the distinctly greener P.Y.194, a benzimidazolone pigment. The fastness to overcoating in this system is perfect. The weatherfastness of these coatings is poor, therefore this pigment grade is mainly used for indoor applications, especially in good value powder coatings, if high lightfastness and weatherfastness are not required.
3.10.5.7 Pigment Orange 61
P.O.61 affords yellowish shades of orange. It is noticeably less fast to organic solvents than comparative yellow types within the same class of pigments.
The paint industry uses P.O.61, like other isoindolinone pigments, to colour high grade industrial finishes, including automotive (OEM) finishes, especially metallic shades. Other areas of application include general industrial and architectural paints and emulsion paints. The pigment is highly lightfast and weatherfast, but does not perform as well as the yellow types within the same class. Incorporated in an alkyd-melamine resin lacquer, the pigment may safely be overcoated up to 140 °C.
P.O.61 is migration resistant in plasticized PVC. Some 1.4% pigment is needed to formulate 1/3 SD colourations containing 5% TiO2. P.O.61 is thus one of the weaker pigments within its range of shades. Its lightfastness is very good. At depths of shade up to 1/25 SD in plasticized PVC, the pigment equals step 7–8 on the Blue Scale. In terms of durability, P.O.61 performs equally well if it is incorporated in PVC plastisols intended for coil coating. This is especially true for transparent samples and those that contain small amounts of TiO2. P.O.61 PVC plastisol systems are heat stable up to 200 °C. Particularly high heat stability is observed in polyolefins. 1/3 SD HDPE samples containing 1% TiO2, for instance, withstand exposure up to 300 °C. P.O.61 produces a shade that is very similar to that of P.O.13. P.O.13 is, on the whole, less fast, but twice as strong tinctorially as P.O.61. P.O.61 causes an uncommon degree of shrinkage in HDPE, which is of great interest to injection moulding. The pigment is recommended as a colourant for polystyrene, polyurethane, unsaturated polyesters and elastomers. High heat stability and good lightfastness make it a suitable candidate for spin dyeing polypropylene and polyacrylonitrile.
The printing ink industry employs P.O.61 mainly in products that are to be printed on PVC films.
3.10.5.8 Pigment Orange 66
P.O.66, a member of the methine series, was introduced some years ago, but has recently already been withdrawn from the market. It was recommended especially for paints. Its colour is a yellowish orange. P.O.66 is a pigment with high tinctorial strength. Excellent lightfastness and weatherfastness qualified the pigment for use in high grade industrial finishes, especially in original automotive and refinishes. The available grade was used in metallic finishes for its high transparency, but was also utilized as a shading pigment. At baking temperatures of 140 °C and higher, P.O.66 is not entirely fast to overcoating. It shows excellent lightfastness in plasticized PVC. P.O.66 dissolves in polyolefins if the temperature exceeds 240 °C.
3.10.5.9 Pigment Orange 69
P.O.69 was introduced to the market some years ago, but meanwhile the marketing has been discontinued. P.O.69 was primarily applied in various types of industrial paints, and the pigment was also recommended for original automotive finishes. The commercial grade, which featured a specific surface area of 30 m2 g−1, has a comparatively coarse particle size. It provides good hiding power. The shade is a yellowish orange. The type was used especially in conjunction with other opaque organic pigments to produce lead-free colourations in the red and brown range. P.O.69 is fast to overcoating up to 140 °C. Both lightfastness and weatherfastness are good, but fail to meet more stringent use requirements. Full shades bleach noticeably and lose much of their gloss.
3.10.5.10 Pigment Red 260
This isoindolinone pigment is already being withdrawn from the market. The commercially available grade had a coarse particle size and thus provided good hiding power. The pigment was recommended for use in various types of industrial paints, including automotive finishes.
P.R.260 may safely be overpainted up to 140 °C. It provides somewhat dull shades in the yellowish red region of the spectrum. P.R.260 is comparatively strong. Full shades and related shades exhibit a slight haze, which is probably due to pigment flocculation. This problem may be met by using suitable additives. Both lightfastness and weatherfastness are good, although full shades darken slightly upon exposure and lose some of their gloss. P.R.260 bleaches considerably in white reductions.
3.10.5.11 Pigment Brown 38
P.Br.38 is already being withdrawn from the market. The commercial type was recommended as a colourant for plastics, especially for PVC and LDPE. The pigment provides yellowish shades of brown and is comparatively strong: 0.64% pigment is needed, for instance, to produce 1/3 SD colourations in plasticized PVC. P.Br.38 shows good migration fastness in plasticized PVC, but is not entirely fast to migration. Its lightfastness in rigid PVC is excellent. Full shade and related shades up to a 1 : 10 TiO2 reduction ratio equal step 8 on the Blue Scale. P.Br.38 is heat stable up to 240 °C, which made it a suitable pigment, especially for LDPE.
Note
References for Chapter 3
- 1 Moser, F.H. and Thomas, A.L. (1983) The Phthalocyanines, vols I and II, CRC Press, Boca Raton, Florida.
- 2 Moser, F.H. and Thomas, A.L. (1963) Phthalocyanine Compounds, Reinhold, New York.
- 3 Booth, G. (1971) The Chemistry of Synthetic Dyes, vol. V (ed. K. Venkataraman), Academic Press, New York, p. 241.
- 4 Lewis, P.A. (ed.) (1988) Pigment Handbook, vol. I, John Wiley & Sons, Inc., New York, p. 679, and further volumes.
- 5 von Braun, A. and Tscherniak, J. (1907) Ber. Dt. Chem. Ges., 40, 2709.
- 6 de Diesbach, H. and von der Weid, E. (1927) Helv. Chim. Acta, 10, 886.
- 7 Hugh, A.G.D., Drescher, A.E., and Thomas, J. (Scottish Dyes Ltd) (1928) GB322169.
- 8 Dandridge, A.G., Drescher, H.A.E., and Thomas, J. (1929) BP 922 169, UK.
- 9 Weissermel, K. and Arpe, H.-J. (1994) Industrielle Organische Chemie, 4th edn, VCH Verlagsgesellschaft, Weinheim.
- 10 Du Pont (1957) US-PS 2 964 532.
- 11 Bayer (1972) DE-AS 2 256 485; BASF (1966) CH-PS 471 865.
- 12 Christie, R.M. and Deans, D.D. (1989) J. Chem. Soc., Perkin Trans II, 193–198.
- 13 Bayer (1974) DE-OS 2 432 564; BASF (1975) DE-AS 1 569 650.
- 14 Erk, P. (2012) personal communication.
- 15 Erk, P. and Hengelsberg, H. (2003) Phthalocyanine dyes and pigments, in Porphyrin Handbook, Volume 19 Applications of Phthalocyanines (eds K.M. Kadish, K.M. Smith and R. Guilard) Elsevier Science, pp. 105–149.
- 16 Horn, D. and Honigmann, B. (1974) XII. Congress FATIPEC, Garmisch, 12–18 May 1974, congress book, pp. 181–187.
- 17 Herbst, W. and Merkle, K. (1970) DEFAZET Dtsch. Farben Z., 24, 365.
- 18 Honigmann, B., Lenné, H.-U., and Schrödel, R. (1965) Z. Kristallogr., 122, 185.
- 19 Booth, G. (1965) Chimia, 19, 201.
- 20 BASF (1980) DE-OS 3 023 722.
- 21 BASF (1978) DE-AS 2 841 244.
- 22 Suzuki, S., Bansho, Y., and Tanabe, Y. (1969) Kogyo Kagaku Zasshi, 72, 720.
- 23 ICI (1960) GB-PS 912 526; ICI (1960) DE-PS 1 161 532.
- 24 BASF (1972) DE-AS 2 210 072.
- 25 Allied (1954) E-AS 1 172 389; Geigy (1963) CH-PS 431 774.
- 26 BASF (1963) DE-PS 1 234 342; Xerox (1970) US-PS 3 492 308; Xerox (1970) US-PS 3 509 146.
- 27 Hunger, K. (ed.) (2003) Industrial Dyes, Wiley-VCH, p. 379.
- 28 BASF (1992) DE-OS 4 234 922.
- 29 Monteath Robertson, J. (1935) J. Chem. Soc., 1935, 615–621.
- 30 Monteath Robertson, J. (1936) J. Chem. Soc., 1935, 1195–1209.
- 31 Linstead, R.P. and Monteath Robertson, J. (1936) J. Chem. Soc., 1936, 1736–1738.
- 32 Erk, P. (BASF) (2002), Communication to the Cambridge Structural Database.
- 33 Hoshino, A., Takenaka, Y., and Miyaji, H. (2003) Acta Crystallogr., Sect. B, 59, 393.
- 34 Brown, C.J. (1968) J. Chem. Soc. A, 1968, 2488–2493.
- 35 Erk, P., Hengelsberg, H., Haddow, M.F., and van Gelder, R. (2004) CrystEngComm, 6, 475–483.
- 36 Dorset, D.L. (1995) Structural Electron Crystallography, Plenum Press, New York, pp. 188–202.
- 36a Erk, P. (BASF) (2001) Personal communication to J. Bernstein.
- 36b Bernstein, J. (2002) Polymorphism in molecular crystals, IUCr Monographs on Crystallography, Clarendon Press, Oxford.
- 36c Janczak, J. (2000) Pol. J. Chem., 74, 157–162.
- 36d Matsumoto, S., Matsuhama, K., and Mizuguchi, J. (1999) Acta Cryst. C, 55, 131–133.
- 36e Zugenmaier, P., Bluhm, T.L., Deslandes, Y., Orts, W.J., and Hamer, G.K. (1997) J. Mater. Sci., 32, 5561.
- 36f Hammond, R.B., Roberts, K.J., Docherty, R., Edmondson, M., and Gairns, R. (1996) J. Chem. Soc., Perkin Trans. 2, 1996, 1527–1528.
- 37 Wynne, K.J. (1984) Inorg. Chem., 23, 4658–4663.
- 37a Czech, C., Kalinowsky, L., and Schmidt, M.U. (2017) Acta Crystallogr., Sect. B, 73, 744–755.
- 38 Hunger, K. (ed.) (2003) Industrial Dyes, Wiley-VCH, pp. 562–569.
- 39 Leznoff, C.C. and Lever, A.B.P. (eds) (1989) Phthalocyanines, vol. 1, vols. 2 and 3 (1993) VCH, New York.
- 40 Kaluza, U. (1979) Physikalisch-chemische Grundlagen der Pigmentverarbeitung für Lacke und Druckfarben, BASF, pp. 90–122.
- 41 Labana, S.S. and Labana, L.L. (1967) Chem. Rev., 67, 1–18; Spengeman, W.F. (1970) Paint Varn. Prod., 60, 37–42.
- 42 Liebermann, H. (1935) Ann. Chem., 518, 245–259.
- 43 Reeve, T.B. and Botti, E.C. (1958) Official Digest, 31, 991–1002.
- 44 Du Pont (1958) US-PS 2 821 529.
- 45 Pollak, P. (1977) Chimia, 30, 357–361; (1977) Progr. Org. Coat., 5, 245; Lonza (1972) DE-OS 2 313 329.
- 46 Du Pont (1958) US-PS 2 821 541; Du Pont (1961) US-PS 3 009 916.
- 47 Du Pont (1961) US-PS 2 969 366.
- 48 Du Pont (1961) US-PS 3 007 930.
- 49 Du Pont (1969) US-PS 3 475 436.
- 50 Allied Chem. (1967) US-PS 3 342 823.
- 51 Urban, M., Schnaitmann, D., and Böhmer, M. (Hoechst) (1996) EP 799862.
- 52 Urban, M., Böhmer, M., Schnaitmann, D., and Weber, J. (Clariant) (1998) EP 971001.
- 53 Kehrer, F. (1974) Chimia, 20, 174–176; Sandoz (1958) DE-PS 1200 457; Sandoz (1962) DE-PS 1 250 033.
- 54 BASF (1963) DAS 1 195 425.
- 55 Du Pont (1958) US-PS 2 844 484.
- 56 Struve, W.S. (Du Pont) (1958), US 2 844 485.
- 57 Jones, F., Okui, N., and Patterson, D. (1975) J. Soc. Dyers Col., 91, 361–365.
- 58 Du Pont (1958) US-PS 2 844 581.
- 59 Jaffe, E.E. (1989) EP 305328.
- 60 BASF (1959) DE 1183884.
- 61 Ciba-Geigy (1986) US-PS 3 074950.
- 62 Jaffe, E.E. (1988) EP 267877.
- 63 Bäbler, F. and Jaffe, E.E. (Ciba-Geigy) (1991), US 748473.
- 64 Toyo (1976) JP Sho51-11372.
- 65 Eastman Kodak (1966) US-PS 3 272 821.
- 66 Tyson, R.S. and Schapiro, L. (1975) DE 2435219.
- 67 Hashizume, K., Miyatake, M., Shigemitsu, M., Kumano, I., Katsura, H., and Oshima, M. (1970) DE 1805266.
- 68 Paulus, E.F., Leusen, F.J.J., and Schmidt, M.U. (2007) CrystEngComm, 9, 131–143.
- 69 Ogawa, T., Furukawa, C., Moriguchi, S., Nemoto, T., Isoda, S., and Kobayashi, T. (1999) Presentation of the 18th Congress of the International Union of Crystallography, Glasgow, 4–13 August 1999, Book of Abstracts, pp. 400–401.
- 70 Gorelik, T.E., Czech, C., Hammer, S.M., and Schmidt, M.U. (2016) CrystEngComm, 18, 529–535.
- 71 Jaffe, E.E. and Chamberlain, T.R. (2009) High Performance Pigments, 2nd edn (eds E.B. Faulkner and R.J. Schwartz), Wiley-VCH, Weinheim, pp. 293–329.
- 72 Paulus, E.F., Dietz, E., Kroh, A., and Prokschy, F. (1989) Presentation on the 12th European Crystallographic Meeting, Moscow, Collected Abstracts Vol. 2, p. 23–24.
- 73 Dietz, E., Kroh, A., Paulus, E.F., Prokschy, F., and Lincke, G. (1991) 11. Internationales Farben-Symposium, Montreux, published in Chimia, 45 (10), 13.
- 74 Nishimura, N., Senju, T., and Mizuguchi, J. (2006) Acta Crystallogr., Sect. E, 62, o4683–o4685.
- 75 Koyama, H., Scheel, H.J., and Laves, F. (1966) Naturwissenschaften, 53, 700.
- 76 Nagai, Y. and Nishi, H. (1968) Senryo Yakuhin, 13, 81–108, p. 90.
- 77 Chung, H. and Scott, R.W. (1971) J. Appl. Crystallogr., 4, 506–511.
- 78 Potts, G.D., Jones, W., Bullock, J.F., Andrewa, S.J., and Maginn, S.J. (1994) J. Chem. Soc., Chem. Commun., 2565–2566.
- 79 Mizuguchi, J., Sasaki, T., and Tojo, K. (2002) Z. Kristallogr. New Cryst. Struct., 217, 249–250.
- 80 Leusen, F.J.J. (1994) Presentation at the 15th European Crystallographic Meeting, Dresden, 1994, Abstract in Z. Kristallogr. Suppl. 8, p. 161.
- 81 Leusen, F.J.J. (1996) J. Crystal Growth, 166, 900–903.
- 82 Lincke, G. (1985) Chem.-Z., 109, 89–96.
- 83 Mizuguchi, J., Senju, T., and Sakai, M. (2002) Z. Kristallogr. New Cryst. Struct., 217, 525–526.
- 84 Otaka, Y. (1975) Nippon Kagaku Kaishi, 1838.
- 85 Senju, T., Nishimura, N., Hoki, T., and Mizuguchi, J. (2005) Acta Crystallogr., Sect. E, 61, o2596–o2698.
- 86 Senju, T., Hoki, T., and Mizuguchi, J. (2005) Acta Crystallogr., Sect. E, 61, o1061–o1063.
- 87 Senju, T., Hoki, T., and Mizuguchi, J. (2006) Acta Crystallogr., Sect. E, 62, o261–o263.
- 88 Labana, S.S. and Labana, L.L. (1967) Chem. Rev., 67, 1–18.
- 89 Ciba Specialty Chemicals (2007) US-PS 2007034117.
- 90 Du Pont (1964) US-PS 3 160 510; Du Pont (1964) US-PS 3 148 075.
- 91 Ciba-Geigy (1982) US-PA 407 644.
- 92 Ciba Holding (1991) EP 466 649; Ciba-Geigy (1992) EP 0466649.
- 93 IntertechPira (2009) The Future of Pigments 2009, 3–5 November 2009, Hamburg, Germany.
- 94 Miles (1991) US-PA 799 453.
- 95 Weiss, C., Wagner, C., Kleimann, C., Rohlfing, M., Tautz, F.S., and Temirov, R. (2010) Phys. Rev. Lett., 105. 086103; Kichin, G., Temirov, R., Wagner, C., and Tautz, F.S. (2012) Nachrichten Chem., 60 (1), 52–54.
- 96 Sandoz (1957) DE-AS 1 071 280.
- 97 Hoechst (1980) DE-OS 3 008 420; Tröster, H. (1983) Dyes Pigments, 4, 171–177.
- 98 BASF (1975) DE-OS 2 545 701; BASF (1975) DE-OS 2 546 266.
- 99 Klebe, G., Graser, F., Hädicke, E., and Berndt, J. (1989) Acta Crystallogr., Sect. B, 45, 69–77.
- 100 Hädicke, E. and Graser, F. (1986) Acta Crystallogr., Sect. C, 42, 189–195.
- 101 Tojo, K. and Mizuguchi, J. (2002) Z. Kristallogr. New Cryst. Struct., 217, 253–254.
- 102 Tojo, K. and Mizuguchi, J. (2002) Z. Kristallogr. New Cryst. Struct., 217, 255–256.
- 103 Tojo, K. and Mizuguchi, J. (2002) Z. Kristallogr. New Cryst. Struct., 217, 45–46.
- 104 Hädicke, E. and Graser, F. (1986) Acta Crystallogr., Sect. C, 42, 195–198.
- 105 Mizuguchi, J. (1998) Acta Crystallogr., Sect. C, 54, 1479–1481.
- 106 Erk, P. (2002) Presentated at Colorchem 02, 9th International Conference on Dyes and Pigments, 12–16 May 2002, Špindlerův Mlýn, Czech Republic.
- 107 Ogawa, T., Kuwamoto, K., Isoda, S., Kobayashi, T., and Karl, N. (1999) Acta Crystallogr., Sect. B, 55, 123–130.
- 108 Kempster, C.J.E. and Lipson, H. (1972) Acta Crystallogr., Sect. B, 28, 3674–3674.
- 109 Mizuguchi, J., Hino, K., and Tojo, K. (2006) Dyes Pigments, 70, 126–135.
- 110 Eckert, W. and Greune, H. (IG Farben) (1924) DRP 430632.
- 111 Fierz-David, H.E. and Rossi, C. (1938) Helv. Chim. Acta 21, 1466–1489.
- 112 Schultz, G. (1931) Farbstofftabellen. 7. Auflage, Akademische Verlagsgesellschaft, Leipzig, Vol. II, p. 132.
- 113 Eckert, W., Greune, H., and Eichholz, W. (IG Farben) (1929) DRP 536911.
- 114 Eckert, W. and Sieber, H. (IG Farben) (1930) DRP 567210.
- 115 FIAT (1948) Final report No. 1313, Vol. II, p. 163–169.
- 116 Schaeffer, A. (1948) Die Küpenfarbstoffe. Aufbau, Eigenschaften Anwendung. Band A: Aufbau der Küpenfarbstoffe. Farbwerke Hoechst, Frankfurt(M)-Höchst, p. 173.
- 117 Weiss, F. (1953) Die Küpenfarbstoffe und ihre Verwendung in der Färberei und im Zeugdruck. Springer-Verlag, Wien, p. 117.
- 118 IG Farben (1932) DE-PS 602 445; Friedländer XX 1433.
- 119 Dietz, E., Kapaun, G., and Schiessler, S. (Hoechst AG) (1989), EP 0367067.
- 120 Lubs, H.A. (Ed.) (1995) The chemistry of synthetic dyes and pigments, Reinhold Publishing Corp., New York, p. 476.
- 121 Mizuguchi, J. (2003) Z. Kristallogr. New Cryst. Struct., 218, 137–138.
- 122 Paulus, E.F. unpublished results.
- 123 Mizuguchi, J. (2003) Z. Kristallogr. New Cryst. Struct., 218, 139–140.
- 124 Teteruk, J.L., Glinnemann, J., Heyse, W., Johansson, K.E., van de Streek, J., and Schmidt, M.U. (2016) Acta Crystallogr., Sect. B, 72, 416–433.
- 124a Paulus, E.F. (1996). Hoechst AG, Internal Report.
- 125 Mizuguchi, J. (2004) J. Phys. Chem. B, 108, 8926–8930.
- 126 DIN 54 071 (1971) Bestimmung der Wetterechtheit von Färbungen and Drucken in Apparaten (künstliche Bewetterung, Xenonbogenlicht).
- 127 Kaul, B.L. (1989) Soc. Plastics Eng., Huron (Ohio), 213–226.
- 128 Farnum, D.G., Mehta, G., Moore, G.G.I., and Siegal, F.P. (1974) Tetrahedron Lett., 15, 2549–2552.
- 129 Yoshida, Z. and Shirota, Y. (eds) (1993) Chemistry of Functional Dyes, vol. 2, Mita Press, Tokyo, pp. 60–67.
- 130 Iqbal, A. et al. (1988) J. Coat. Technol., 60, 37–45.
- 131 Ciba-Geigy (1983) US-PS 4 415 685.
- 132 Smith, H.M. (ed.) (2002) High Performance Pigments, Wiley-VCH Verlag GmbH, pp. 167–170.
- 133 Ciba-Geigy (1994) EP 690 058 A1.
- 134 Mizuguchi, J., Grubenmann, A., Wooden, G., and Rihs, G. (1992) Acta Crystallogr., Sect. B, 48, 696–700.
- 135 Mizuguchi, J., Grubenmann, A., and Rihs, G. (1993) Acta Crystallgor., Sect. B, 49, 1056–1060.
- 136 Mizuguchi, J. and Miyazaki, T. (2002) Z. Kristallogr. New Cryst. Struct., 217, 43–44.
- 137 Mizuguchi, J. and Shikamori, H. (2003) Z. Kristallogr. New Cryst. Struct., 218, 127–128.
- 138 Mizuguchi, J. and Shikamori, H. (2004) J. Phys. Chem. B, 108, 2154–2161.
- 139 Mizuguchi, J. (1998) Acta Crystallogr., Sect. C, 54, 1482–1484.
- 140 Mizuguchi, J. and Matsumoto, S. (2000) Z. Kristallogr. New Cryst. Struct., 215, 195–196.
- 141 Baeyer, A. and Emmerling, A. (1870) Ber. Dtsch. Chem. Ges., 3, 514–517.
- 142 Susse, P., Steins, M., and Kupcik, V. (1988) Z. Kristallogr., 184, 269–273.
- 143 Zollinger, H. (2003) Colour Chemistry, Wiley-VCH, Weinheim.
- 144 Hoechst (1983) DE-OS 3 324 879.
- 145 Bayer (1974) DE-OS 2 457 703.
- 146 Hoechst (1975) DE-OS 2 504 962.
- 147 Hoechst (1975) DE-OS 2 043 820.
- 148 BASF (1979) DE-OS 2 916 400.
- 149 Fukushima, K., Nakatsu, K., Takahashi, R., Yamamoto, H., Gohda, K., and Homma, S. (1998) J. Phys. Chem. B, 102, 5985–5990.
- 150 Kaul, B.L. (1971) CH 549626.
- 151 Buchsbaum, C., Paulus, E.F., and Schmidt, M.U. (2011) Z. Kristallogr., 226, 822–831.
- 152 Senju, T. and Mizuguchi, J. (2003) Z. Kristallogr. New Cryst. Struct., 218, 129–130.
- 153 Woroshzow, N.N. (1966) Grundlagen der Synthese von Zwischenprodukten und Farbstoffen, 4th edn, Akademie-Verlag, Berlin.
- 154 Bien, H.-S. et al. (1985) Ullmann's Encyclopedia of Industrial Chemistry, 5th edn, vol. A2, VCH, Weinheim.
- 155 BASF (1965) DE-AS 1 544 372; BASF (1965) DE-OS 1 544 374.
- 156 BASF (1970) DE-OS 2 059 677.
- 157 Kanter, H. and Ort, B. (1986) Presented at XVIII Congrress FATIPEC, 21–26 September 1986, Venice, Italy, Congress book, vol. 3, pp. 95–111.
- 158 Bayer (1976) DE-OS 2 644 265.
- 159 Manukian, B.K. and Mangini, A. (1971) Helv. Chim. Acta, 54, 2093–2097.
- 160 Ciba (1960) CH-PS 396 264.
- 161 Ogawa, K., Scheel, H.J., and Laves, F. (1966) Naturwissenschaften, 53, 700–701.
- 162 Ogawa, K. and Scheel, H.J. (1969) Z. Kristallogr., 130, 405–419.
- 163 Interchemical (1953) US-PS 2 727 044.
- 164 Ciba (1962) DE-PS 1 283 542; Ciba (1967) DE-AS 1795 102.
- 165 Sabnis, R.W. (1999) Displays, 20, 119; Iwasaki, M. (1998) Kinosei Ganryo Gijutsuto Oyo Tenkai, Tokio, p. 188.
- 166 Kiel, E.G. and Heertjes, P.M. (1963) J. Soc. Dyers Col., 79, 21–27.
- 167 BASF (1973) DE-OS 2 300 019.
- 168 Rys, P. and Zollinger, H. (1982) Farbstoffchemie, 3rd edn, Verlag Chemie, Weinheim, p. 146; Zollinger, H. (1991) Colour Chemistry, 2nd edn, VCH, Weinheim.
- 169 Venkataraman, K. and Iyer, V.N. (1971) The Chemistry of Synthetic Dyes, vol. V, (ed. K. Venkataraman), Academic Press, New York, pp. 182–236.
- 170 For example, BASF (1978) DE-AS 2 854 190.
- 171 Ciba-Geigy (1970) DE-OS 2 105 286.
- 172 Ciba-Geigy (1977) DE-OS 2 812 192.
- 173 BASF (1977) DE-AS 2 748 860.
- 174 Allied Chem. Corp. (1973) DE-OS 2 428 121.
- 175 Kunz, M.A. (1939) Angew. Chem., 52, 269–282.
- 176 British Intelligence Objectives Sub-committee (1946) BIOS Report. German Organic Pigments and Lake Dyestuffs, British Intelligence Objectives Sub-committee. London, H.M. Stationary Office. Technical information and documents unit, London 1946 (Investigations 24 July to 23 August 1946), pp. 412–463. This is a translation of an unpublished report on the application of the X-ray technique in the chemical industry by Dr. Georg von Susich of Wilhelmsfeld near Heidelberg relating to the work of von Susich at the I.G. laboratories in Ludwigshafen (1934).
- 177 Field Information Agency, Technical (FIAT) (1948) Final Report No. 1313, German Dyestuffs and Dyestuff Intermediates, including Manufacturing Processes, Plant Design, and Research Data, vol. 3: Dyestuff Research, prepared by Field Information Agency, Technical (FIAT), United States Group Control Council for Germany (1948).
- 178 Dai Nippon Ink (1998) US 5788759.
- 179 BASF (1977) DE-AS 2 705 107.
- 180 DE-AS 28 541 900.
- 181 Bailey, M. (1955) Acta Crystallogr., 8, 182–185.
- 182 Stadler, H.P. (1953) Acta Crystallogr., 6, 540–542.
- 183 BASF (1970) DE-AS 2 007 848.
- 184 BASF (1977) DE-AS 2 702 596.
- 185 Schmidt, M.U., Paulus, E.F., Rademacher, N., and Day, G.M. (2010) Acta Crystallogr., Sect. B, 66, 515–526.
- 186 BASF (1961) DE-AS 1 284 092.
- 187 BASF (1979) DE-AS 2 909 568.
- 188 Bien, H.-S., Stawitz, J., and Wunderlich, K. (2005) Anthraquinone Dyes and Intermediates, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH.
- 189 Bios Final Report No. 987, page 75.
- 189a Bien, H.-S., Stawitz, J., and Wunderlich, K. (2005). Anthraquinone Dyes and Intermediates, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim.
- 189b Howell, E.T. (Du Pont) (1944) US 2388743.
- 189c Lüttringhaus, A., Neresheimer, H., and Emmer, H.J. (IG Farben) (1925) DE 450999.
- 189d Bolton, W. and Stadler, H.P. (1964) Acta Cryst. 17, 1015–;1020.
- 190 Stedlmayr, R. (Hoechst) (1952) DE 946 560.
- 191 Arndt, O. and Papenfuhs, T. (Hoechst) (1988) DE-OS 3 707 148; Desmurs, J.-R. and Jouve, I. (Rhone-Poulenc) (1988) EP 326 455 and EP 326 456.
- 192 British Intelligence Objectives Sub-committee (1945) BIOS Final Report No. 960, London, H.M. Stationary Office, p. 75.
- 193 Hetschko, M. and Hufnagel, T. (Cassella) (1980) DE-OS 3 010 949.
- 194 Schmidt, M.U., Ermrich, M., and Dinnebier, R.E. (2005) Acta Crystallogr., Sect. B, 61, 37–45.
- 195 Schmidt, M.U., Kempter, P., Plüg, C., and Born, R. (Clariant) (2002) EP 1201718.
- 196 Pugin, A. (1965) Offic. Dig. Fed. Soc. Paint Technol., 37, 782–802.
- 197 Schmidt, M.U., Kempter, P., and Born, R. (Clariant) (2002) EP 1199309.
- 198 von der Crone, J. and Pugin, A. (Ciba) (1962) DE 1 243 303.
- 199 Frey, C., Mory, R., and Moser, E. (Ciba) (1959) DE 1 142 212.
- 200 Ronco, K. (Ciba) (1959) DE 1 174 927; Frey, C., Grieder, E., and Ronco, K. (Ciba) (1961) DE-AS 1 195 005.
- 201 Kempter, P. and Wilker, G. (2001) Farbe Lack, 107, 29–31.
- 202 Hunger, K. (ed.) (2003) Industrial Dyes, Wiley-VCH Verlag GmbH, p. 109, 139; Manukian, B.K. and Mangini, A. (1970) Chimia, 24, 328–339.
- 203 Patil, A., Patkar, G., and Vaidyanathan, T. (1974) Bombay Technol., 24, 26–33.
- 204 BASF (1968) DE-AS 1 770 960.
- 205 Sakai, H., Ando, H., Murata, H., Aoki, S., and Oshiumi, I. (1982) J. Jpn. Soc. Colour Mater., 55, 685.
- 206 Fujii, I., Kodama, T., Yanagihara, N., and Hirayama, N. (2004) Anal. Sci., 20, x35–x36.
- 207 BASF (1990) DE-OS 4 020 423.
- 208 Teijin Ltd. (1976) DE-AS 2 706 872.
- 209 Ciba (1964) CH-OS 438 542.
- 209a Gumbert, S.D., Körbitzer, M., Alig, E., Schmidt, M.U., Chierotti, M.R., Gobetto, R., Li, X., van de Streek, J. (2016) Dyes & Pigments, 131, 364–372.
- 210 Pugin, A. and Crone, J.v.d. (1966) Farbe Lack, 72, 206–217.
- 211 Sandoz (1972) DE-OS 2 322 777.
- 212 Ciba-Geigy (1990) EP-PA 479 727.
- 213 Dainippon Ink (1973) DE-OS 2 321 511.
- 214 Dainichi Seika (1975) JA-PS J 5 1088 516; JA-PS J 5 2005 840; Dainichi Seika (1975) J 5 2005 841.
- 215 Toyo Soda (1978) JA-PS J 5 5012106.
- 216 Dainichi Seika (1978) JA-PS J 5 5034 268; Ciba-Geigy (1973) DE-OS 2 438 867; Sumitomo (1970) JA-PS 7 330–659/658; Ciba-Geigy (1976) CH-PS 613 465; BASF (1979) DE-OS 2 909 645.
- 217 Ciba-Geigy (1975) DE-OS 2 548 026.
- 218 Ciba-Geigy (1975) DE-OS 2 606 311.
- 219 Ciba-Geigy (1979) EP-PS 29 413.
- 220 Ciba-Geigy (1974) DE-OS 2 518 892.
- 221 Ciba-Geigy (1976) DE-OS 2 733 506.
- 222 Dainichi Seika (1978) DE-OS 2 901 121.
- 223 Ciba-Geigy (1979) EP-PS 22 076.
- 224 Erk, P., Hengelsberg, H., Haddow, M.F., and van Gelder, R. (2004) CrystEngComm, 6, 474–483.224avan de Streek, J., Ivashevskaya, S.N., and Schmidt, M.U. (2018) in preparation.
- 225 BASF (1979) DE-AS 2 914 086; BASF (1980) EP-PS 35 672; Ciba-Geigy (1979) EP 29 007.
- 226 Crone, J.v.d. (1985) J. Coat. Technol., 57 (725), 67–72.
- 227 Ciba-Geigy (1977) DE-OS 2 814 526.
- 228 BASF (1976) DE-OS 2 628 409; BASF (1970) DE-OS 2 041 999.
- 229 Bayer (1980) DE-OS 3 022 839.
- 230 Bayer (1989) DE-OS 3 935 858.
- 231 BASF (1977) DE-OS 2 757 982.
- 232 Bayer (1991) EP 510 436.
- 233 Ciba-Geigy (1978) DE-OS 2 924 142; Ciba-Geigy (1979) EP-PS 19 588.
- 234 Radtke, V., Erk, P., and Sens, B. (2009) High Performance Pigments, 2nd edn (eds E.B. Faulkner and R.J. Schwarz), Wiley-VCH Verlag GmbH, Weinheim, pp. 221–242.
- 235 Lewis, P.A. (ed.) (1988) Pigment Handbook, vol. 1, John Wiley & Sons, Inc., New York, pp. 713–722.
- 236 Kaul, B.L. (1993) Rev. Prog. Colour., 23, 19–35.
- 237 Geigy (1956) DE-PS 1 098 126; Ciba-Geigy (1977) DE-OS 2 804 062.
- 238 Sakai, I.d. et al. (1982) J. Jpn. Soc. Col. Mater., 55, 683.
- 239 Bayer (1980) DE-OS 3 022 839.
- 240 Kurtz, W. (BASF) (1982) Symposium Druckfarbenindustrie, Ludwigshafen, Germany, p. 31.
- 241 Wörth, H. (1988) Presented at XIX Congress FATIPEC, 19–21 September 1998, Aachen, Congress book, vol. 4, pp. 49–65.
- 242 BASF (1970) DE-OS 2 041 999.
- 243 (a) Schädeli, U., Tingueli, E., Hall-Goulle, V., and Keyzer, G. (CIBA SC), “Black-pigmented structured high molecular weight material”, WO1998/045757, Example 5. (b) Maurer, M. and Raetzko, N. (CIBA), “New C.I. Pigment Violet 37 in rod-like form”, WO 2008/071585A2.