
7
Multivalency as a Design Criterion in Catalyst Development
Paolo Scrimin, Maria A. Cardona, Carlos M. León Prieto, and Leonard J. Prins
Department of Chemical Sciences, University of Padova, Via Marzolo 1, 35131, Padova, Italy
7.1 Introduction
Multivalency is most often associated with the binding interaction between molecular partners through the simultaneous occurrence of multiple binding events. The aim of this chapter is to illustrate how multivalency relates to catalysis, in particular referring to those cases in which multivalency is purposely used as a design criterion to develop catalysts. The attachment of homogenous catalysts to multivalent scaffolds such as dendrimers, nanoparticles, or macroscopic resins has received tremendous attention in the past decades [1–5]. This interest is predominantly caused by the possibility to create hybrid systems that combine the advantages of heterogeneous and homogeneous catalysts [6]. Anchoring of a catalyst on a solid support creates the possibility of catalyst separation and, thus, recycling, which leads to a potential cost reduction. Although of obvious importance, multivalent catalysts that have been prepared for this purpose will not be discussed here and the reader is referred to the numerous reviews cited above that provide overviews of such systems. Rather, in this chapter the focus will be on systems in which multivalency is an essential prerequisite for observing or enhancing catalytic activity. This will involve a discussion of synthetic systems that express cooperative catalysis. This implies that, just as what happens in the active site of an enzyme, catalytic activity originates from the interplay between two (or more) functional groups that are in close proximity in the multivalent scaffold. Indeed, special attention will be paid to the interpretation of the Michaelis–Menten parameters for such multivalent enzyme mimics. Additional topics that will be treated are the ability of the multivalent system to alter the local reaction conditions and induce different reaction mechanisms. Finally, a section is dedicated to what happens in the special case where a multivalent catalyst acts on a multivalent substrate. It is not the purpose of this chapter to provide an exhaustive overview of all examples that have appeared in the literature. Examples have been selected based on the insight they can provide in discussing the relationship between multivalency and catalysis.
7.2 Formation of Enzyme‐Like Catalytic Pockets
The close proximity of multiple functional groups in a single molecular structure provides multivalent systems with an excellent opportunity to create catalytic sites similar to the active site of enzymes. The emergence of dendrimers led rapidly to the realization that the dendritic shell may have a strong effect on the performances of a catalytic core. As illustration we report an example by Javor et al. [7], who have extensively studied peptide dendrimers as enzyme‐like catalysts [8,9]. The attractiveness of this system is the simplicity of the catalytic site in the core and the enormous structural variety inserted in the branches of the third generation dendrimer (Figure 7.1). The fact that amino acids are used to construct the structure strongly enhances the analogy with enzymes. A library of over 65 000 peptide dendrimers was prepared following a combinatorial approach. Variable amino acids at the catalytic core included nucleophilic (His, Cys) and cationic (Arg) residues for substrate binding and catalysis. In the outer regions, aromatic residues were chosen between aromatic residues (Tyr, Phe, Trp) to assist in binding. Finally, polar, negatively charged residues and small hydrophobic residues were distributed throughout the entire structure. The library was screened on‐bead for esterase activity using a fluorogenic butyrate ester. Strongly fluorescent beads were then selected for sequencing and analysed for the presence of consensus‐sequences. Active sequences were found to contain at least one histidine or arginine in the catalytic core and predominantly aromatic residues at the outer positions. Representative hits were resynthesized and were found to catalyse the hydrolysis of activated esters with saturation kinetics and multiple turn‐overs. An important contribution to catalysis by the apolar outer layers was observed attributed to an increase in substrate binding. The importance of this study is that it demonstrates the possibility to create multivalent enzyme‐like structures in which the properties of the active site are altered because of the surrounding structure.

Figure 7.1 On‐bead selection of a catalytic dendrimer from a combinatorial library of 65 536 different dendrimers. The yellow circle highlights the reactive core with a substrate.
Source: Ref. [7]. Reproduced with permission of American Chemical Society.
The attractive feature of dendrimers is their monodisperse structure, which is well‐defined on the molecular level. This allows for a precise determination of the effect of structural changes on the catalytic performances of the system. Yet, the multi‐step covalent synthesis of dendrimers poses challenges in terms of yields and purification. For that reason, self‐assembled monolayers (SAMs) on gold nanoparticles have emerged as attractive alternatives for the formation of multivalent catalytic systems, because they form spontaneously via a strong sulfur–gold (Au) interaction [10,11]. The potential of these systems to create enzyme‐like catalysts was nicely illustrated by Belser et al. [12] in a study on catalytic SAMs on gold nanoparticles. They distinguished two different situations for a mixed monolayer system composed of two different thiols, one bearing a catalytic group and the other one with an inert head group (Figure 7.2). A convex catalytic site is formed in the case where the catalytic thiol contains a longer spacer compared with the surrounding inert thiol. In that case, the catalytic group extends out of the monolayer surface. In principle, the activity should resemble that of the monomeric reference catalyst, although the neighbouring head groups of inert thiols may affect the catalytic activity because of interactions with the substrate or an alteration of the local chemical environment (see examples below). Alternatively, in the case where the catalytic thiol has a shorter spacer compared with the surrounding thiol a concave catalytic site is formed, which has more similarities with the active site of an enzyme. In an initial study, they explored the first case by embedding thiolates with chiral rhodium‐PYRPHOS head groups in monolayers of n‐alkanethiolates of different length and head group polarity on Au nanoparticles (NPs) with a diameter of around 3 nm. The catalytic activity of these systems was evaluated in the hydrogenation of methyl α‐acetamido‐cinnamate. Scanning tunnelling microscopy measurements of analogously prepared two‐dimensional (2D) monolayers revealed a statistical distribution of the catalytic head groups, which is of crucial importance when the synergetic effect between the two thiols is studied (see Section 7.5.2). As expected, the catalytic activity was similar to that of the analogous homogeneous catalyst both in terms of enantioselectivity and conversion, but only in the case where apolar head groups were present on the neighbouring thiols. The presence of polar amino and hydroxy end groups caused a decisive decrease in yield and enantioselectivity. Although this study did not provide an explanation for this effect, it clearly shows that neighbouring end groups play an important role in governing the performance of the catalytic system.

Figure 7.2 Formation of convex and concave catalytic sites on a monolayer depending on the relative spacer length of the catalytic and surrounding inert thiols.
Source: Ref. [12]. Reproduced with permission of American Chemical Society.
A more complete demonstration was provided by Paluti and Gawalt [13,14] in a study on the activity of aza‐bis(oxazoline) copper complexes embedded in 2D SAMs composed of alkane thiols (Figure 7.3). Apart from the convex situation analysed by Belser et al., systems were also analysed in which the catalyst was embedded within the monolayer or at an equal distance compared with the surrounding head groups. All systems were tested in the cyclopropanation reaction of ethyl diazoacetate and styrene, thus permitting an analysis of the product distribution both in terms of cis/trans ratio as well as the enantioselectivity of each diastereoisomer. As a reference, the monomeric aza‐bis(oxazoline) catalyst with bulky t‐butyl substituents gave a cis/trans ratio of 20/80 with respective enantiomeric excess (ee) values of 80 and 87%. Nearly identical results were obtained in the case where the catalyst was positioned well above the surrounding monolayer (cis/trans 23/77; eecis = 81; eetrans = 85) indicating that this construction provides supported catalysts with homogeneous properties. Changes were observed upon positioning the catalyst at an even distance compared with the monolayer surface. The cis/trans ratio changed slightly to 16/84, but a significant increase in the ee of the trans product was observed (from 87 to 93%). For the cis isomer no change in enantioselectivity was reported (82 versus 80), but, remarkably, the opposite enantiomer was favoured. Finally, embedding the catalyst within the monolayer caused a drop both in the cis/trans ratio (28/72), but also in the ee values of both products (eecis = 37; eetrans = 44). The latter results were tentatively ascribed to the occurrence of steric interactions between the alkyl chains and the catalysts. From this comparative study it emerges that the most advantageous situation occurs when the catalyst is levelled with the monolayer surface.

Figure 7.3 Three catalytic 2D SAMs used to study the cyclopropanation reaction between styrene and ethyl diazoacetate.
Source: Ref. [13]. Reproduced with permission of Elsevier.
7.3 Cooperativity Between Functional Groups
An important step towards artificial enzymes requires the exploitation of multivalent scaffolds for the formation of catalytic sites in which multiple functional groups cooperatively act on the substrate [15]. This way the scaffold assumes a fundamental role in preorganizing the chemical functionalities. Typically, this results in very strong rate accelerations over the background, because the monomeric units by themselves are not or hardly active. A classic example of cooperative catalysis is the imidazole‐catalysed hydrolysis of carboxylic esters around pH 7 in which different imidazoles provide both nucleophilic and a general acid/base contribution. Delort et al. [16] explored the occurrence of this mechanism in a series of peptide dendrimers of different generation (up till the fourth) containing His‐residues in every generation (Figure 7.4). An additional Ser‐residue was also presented in each generation as previous studies had shown an enhanced activity when this residue was present. Importantly, all dendrimers exhibited enzyme‐like saturation kinetics in the hydrolysis of pyrene trisulfonate esters and Michaelis–Menten parameters could be determined for each generation. Comparison of the kcat, KM and kcat/KM values for each generation gave a valuable insight in the cooperativity between functional groups in the dendrimer. A systematic study of the dendritic effect in peptide dendrimer catalysis revealed that the catalytic rate constant kcat and substrate binding constant 1/KM both increased with increasing generation number. The dendrimers showed rate accelerations up to kcat/kuncat = 20 000 and KM values around 0.1 mM. The experiments showed thus a strong positive dendritic effect resulting from cooperative binding and catalysis [17–20]. A very strong indication for the effective occurrence of cooperative interactions between imidazoles came from the very different rate profiles as a function of pH compared with the monomeric reference catalyst 4‐methylimidazole. For the reference catalyst, an increase in rate was observed as the pH increased in agreement with a deprotonation of the imidazole (creating the nucleophile). On the other hand, for the dendrimer‐catalysed reactions the rate was slightly bell‐shaped over the pH range studied (4.5–7.5) indicating a double role of the imidazole in the mechanism.


Figure 7.4 Catalytic dendrimers containing His‐residues in every generation. Observed positive dendritic effect in the hydrolysis of activated esters.
Source: Ref. [16]. Reproduced with permission of American Chemical Society.
Another class of reactions that generally requires the joined action of two catalytic groups is the hydrolytic cleavage of phosphodiesters [21,22]. This reaction is biologically very relevant as phosphodiesters constitute the backbone of DNA and RNA, the polymers that carry genetic information. The high stability of this bond is reflected by the estimated half‐life of 1010 years for the cleavage of dimethyl phosphate. Not far from this low reactivity is the time required for hydrolytically cleaving the P‐O bond of DNA. On the other hand, RNA is more labile because the nucleophilic attack on phosphorus is performed intramolecularly by the ‐O(H) in the 2'‐position of the ribose. This reduces the half‐life of RNA to roughly 104 years. Enzymes involved in the DNA or RNA cleavage typically have multiple transition metal ions (Zn2+, Mg2+) in the active site. These metal ions work in a concerted manner through nucleophile activation and stabilization of both the transition state and leaving group. Accordingly, this reaction provides an excellent test case to determine the occurrence of cooperativity in multivalent catalysts. Manea et al. [23] prepared Au NPs with a mixed monolayer composed of thiols terminating with a catalytic 1,4,9‐triazacyclonanone (TACN) · Zn2+ head group and inert octane thiols (Figure 7.5). NMR spectroscopic analysis revealed that the thiols were present in a 1.2:1 ratio in the monolayer. The catalytic activity of the NPs was tested on the transphosphorylation of 2‐hydroxypropyl‐p‐nitrophenyl phosphate (HPNPP), which is typically used as an RNA model compound. Importantly, Zn2+ is fundamental to catalysis, because hardly any activity over the background reaction is observed in the absence of metal ion. This offers an attractive possibility to correlate the presence of active catalysts on the surface to the overall activity by measuring the reaction rate as a function of the amount of Zn(NO3)2 added to a NP solution. The observed sigmoidal profile is a strong indication that the catalytic activity originates from the cooperative action of two neighbouring TACN · Zn2+ complexes (Figure 7.5). At low Zn2+ concentrations isolated complexes are formed in the monolayer with a low catalytic activity. However, after a TACN:Zn2+ ratio of around 1:0.3 has been reached a strong increase in reactivity is observed, because the gradual saturation of the monolayer with Zn2+ causes the formation of dinuclear catalytic sites. A maximum reactivity of the NPs is reached when the monolayer is fully saturated with Zn2+. In subsequent studies, this observation of cooperativity was used to understand in detail the origin of the dendritic effect in multivalent catalysts (see Section 7.5).

Figure 7.5 Catalytic NPs for the transphosphorylation of HPNPP, an RNA model substrate. A sigmoidal curve is observed when the reaction rate is measured as a function of the number of equivalents of Zn2+ (relative to the head groups).
Source: Ref. [23]. Reproduced with permission of John Wiley and Sons.
The induction of cooperativity between neighbouring groups in multivalent systems is not limited to biomimetic reactions, but is an attractive strategy for any reaction that has an order higher than one in the concentration of catalyst. A well‐known example is the asymmetric ring opening of epoxides catalysed by chiral salen · Co3+ complexes. Substantial mechanistic evidence is in support of a mechanism involving the cooperative action of two complexes through the simultaneous activation of both the nucleophile and the epoxide (Figure 7.6a and b) [24,25]. Early on, this led to the speculation that the incorporation of these complexes in dendritic structures might lead to highly efficient catalytic systems because of an enforced cooperativity [26]. Commercially available polyamidoamine (PAMAM) dendrimers containing 4, 8 or 16 head groups were functionalized at the periphery with chiral salen · Co3+ complexes and tested for catalytic activity in the hydrolytic kinetic resolution of a terminal epoxide (Figure 7.6c). Not only did the catalytic dendrimers exhibit a significantly enhanced activity (normalized for the number of Co3+ complexes) compared with the monomeric complex, but, surprisingly also compared with the reference dimeric complex. Interestingly, a maximum reactivity per cobalt was attained for the dendrimer containing four complexes. The positive dendritic effect was ascribed to restricted conformations imposed by the dendrimer structure, creating a higher effective molarity of salen · Co3+ complexes. An alternative explanation relied on the occurrence of higher order interactions between the catalytic centres in dendrimers of higher generation. Later on (Section 7.4), an example will be discussed in which the multivalent structure indeed induces a different mechanistic pathway, which is one of the hallmarks of enzymatic catalysis. The same catalytic units were also exploited in a Au NP‐based multivalent catalyst by inserting salen‐terminated thiols in an n‐octanethiol covered monolayer through the place exchange reaction [27]. A final 3:1 ratio of catalytic and inert thiolates ensured the possibility of forming dinuclear catalytic pockets. The observation that this NP exhibited a complete kinetic resolution of racemic hexane‐1‐oxide within just 5 h (as compared with 52 h required for the monomeric reference catalyst at the same loading) confirmed the efficacy of embedding this catalyst on a NP.

Figure 7.6 (a) Cooperative action of two salen · Co3+ complexes. (b) The hydrolytic ring‐opening of epoxides. (c) Example of a PAMAM dendrimer functionalized with eight chiral salen · Co3+ complexes.
Source: Ref. [26]. Reproduced with permission of John Wiley and Sons.
7.4 Mechanistic Effects
Apart from a direct control over activity by creating catalytic sites through the precise positioning of functional groups on a multivalent scaffold, it has also been demonstrated that the scaffold itself can exert an indirect effect on catalysis by creating a local chemical environment that is different from the bulk. This is exemplified by a study of Au NPs terminating with a HisPhe‐OH dipeptide (Figure 7.7) [28]. The monomeric peptide itself is a modest catalyst for the hydrolysis of 2,4‐dinitrophenylbutanoate. Its incorporation in the nanosystem led to a significant increase in activity of at least one order of magnitude. Yet, the most interesting difference was the observed catalytic activity as a function of pH. For the monomeric peptide, an increase in activity was observed upon an increase in pH, consistent with the deprotonation of imidazole (pKa = 6.6), which is the catalytically relevant nucleophile. On the other hand, the profile observed for the NPs indicates the formation of the first nucleophilic species with pKa 4.2 and a second one with pKa 8.1. These pKa values were assigned to the carboxylic acid and the imidazolium, respectively. The reason for the higher value of the pKa of the imidazolium in the NP is due to the anionic nature of the NP that disfavours the deprotonation of the imidazolium cation. The confinement of the catalytic units in the monolayer covering the NPs triggers a cooperative hydrolytic mechanism operative at pH < 7 in which a carboxylate and an imidazolium ion act as a general base and general acid, respectively. The absence of this mechanism in the monomeric catalyst results in a 300‐fold rate acceleration at acidic pH for the NP‐based catalyst.

Figure 7.7 Catalytic activity of Au NPs functionalized with a catalytic dipeptide as a function of pH compared with that of the monomeric reference catalyst.
Source: Ref. [28]. Reproduced with permission of American Chemical Society.
In an entirely different system enhanced catalytic activity was also found to originate from a local pH effect in combination with other factors. Zaramella et al. [29,30] self‐assembled small negatively charged peptides on the surface of Au NPs passivated with thiols containing positively charged quaternary ammonium salts (Figure 7.8). These peptides were equipped with one or more His‐residues as catalytic units for the hydrolysis of esters. When bound to the surface, the peptides accelerated the cleavage of the p‐nitrophenyl ester of N‐Cbz‐protected phenylalanine by more than two orders of magnitude. However, this rate enhancement did not originate from the cooperative action between two His‐residues on the same peptide because a linear correlation was observed between the number of His‐residues present in the peptides and the second‐order rate constant. Yet, cooperativity was not observed between His‐residues on neighbouring peptides, because in that case the rate should have exponentially increased upon saturating the monolayer surface with peptides. A detailed analysis showed that the main reason for the enhanced catalytic activity was the co‐localization of substrate and catalyst on the multivalent surface. Importantly, the catalysis was further enhanced by the local pH at the surface which was found to be 0.7 units higher than the pH of the bulk solvent, caused by the cationic ammonium groups. The pH tuning by the charge of the surface is very similar to what is observed with cationic micelles or vesicles. The higher local pH increased the concentration of unprotonated imidazole which acted as the nucleophile during catalysis.

Figure 7.8 Catalytic activity caused by the co‐assembly of substrate and catalyst on a Au NP surface aided by an increase in local pH.
Source: Ref. [29]. Reproduced with permission of American Chemical Society.
7.5 The Dendritic Effect in Multivalent Nanozymes
Synthetic multivalent catalysts frequently display enzyme‐like Michaelis–Menten reaction kinetics and have been coined nanozymes also for that reason [23,31]. The basis of Michaelis–Menten kinetics is a model that assumes that the substrate (S) is bound by an enzyme (E) yielding the complex E · S, after which the chemical transformation into product (P) takes place. Complex formation is determined by the dissociation constant KM, whereas the efficiency of the catalytic process is determined by the first‐order rate constant kcat. This leads then to the following expression for the initial rate vinit.

The values for KM and kcat are obtained by fitting a plot of the initial rate as a function of the initial substrate concentration to Equation 7.1. As mentioned, multivalent catalysts frequently display similar saturation kinetics and, consequently, the catalytic properties of such systems are typically evaluated in terms of the parameters kcat and KM. Comparison of these values with those of the monomeric reference catalyst or comparison of these values within a series of structurally multivalent catalysts, such as dendrimers of different generations, has often led to the observation that multivalent catalysts display a strong dendritic effect. This implies that the catalytic performances progressively increase as a function of the valency of the scaffold.
Yet, an intrinsic difference exists between an enzyme and the multivalent catalysts described in this chapter. The Michaelis–Menten model at the basis of Equation 7.1 explicitly refers to an enzyme with a single active site (Figure 7.9). This implies that the enzyme forms a 1:1 complex with the substrate and that, consequently, at saturation each enzyme is saturated with a single substrate. Evidently, this is not the case for multivalent catalysts. Here, multiple binding sites are present and at saturation the single multivalent catalyst is saturated with multiple substrates (Figure 7.9). Thus, the kcat and KM values obtained from fitting the saturation curve to Equation 7.1 are macroscopic values composed of all microscopic binding and catalytic events that occur simultaneously within the multivalent system. For two multivalent systems, a dendritic catalyst and a Au NP‐based catalyst, our group has developed theoretical models in order to determine the relation between the microscopic values for the individual catalytic sites and the macroscopic ones measured for the entire system [32,33]. These studies give a surprising insight into the origin of the dendritic effect.

Figure 7.9 Michaelis–Menten saturation kinetics for an enzyme and a multivalent nanozyme.
7.5.1 Peptide‐Based Dendrimers for the Cleavage of Phosphodiesters
A series of dendrimers of different generation with a peptidic Lys‐backbone and a varying number of peripheral 1,4,9‐triazacyclononane (TACN) · Zn2+ complexes (ranging from 4 to 32) were analysed for their ability to catalyse the transphosphorylation of HPNPP (Figure 7.5 and Figure 7.10) [32]. Michaelis–Menten‐like saturation kinetics was observed and values for kcat and KM were obtained by fitting the data to Equation 7.1. Plots of the obtained values as a function of the valency indicate an increase of kcat and a decrease in KM (stronger binding) as the valency of the dendrimer increases (Figure 7.10). At first glance, this suggests that a positive dendritic effect originates from both an improved catalytic efficiency and a higher affinity of the substrate for the catalyst. The combined effect leads to a significant increase in the apparent second‐order rate constant kcat/KM, often taken as a measure to quantify the dendritic effect. Yet, how do these results need to be interpreted?

Figure 7.10 Representative structure of a peptidic Lys‐based dendrimer with 32 catalytic TACN•Zn2+ head groups and plots of the overall Michaelis–Menten parameters for the cleavage of HPNPP (see Figure 7.5) as a function of the number of catalytic units present in the dendrimer.
Source: Ref. [32]. Reproduced with permission of American Chemical Society.
Obviously, because of the presence of multiple catalytic head groups in the dendrimers the value for kcat is artificially inflated. A correct comparison of kcat between the different generations requires a normalization on the actual number of catalytic head groups present. Indeed, after correction it is observed that the kcat value increases only from dendrimer D4 to D8 after which it remains constant and even drops for the D32 (D4, D8 and D32 refer to dendrimers with 4, 8 or 32 TACN head groups, respectively). This indicates that from an efficiency point of view, the catalytic process does not improve upon increasing the valency of the dendrimer. A less obvious issue that needs to be considered in evaluating kcat is the maximum saturation level that can be reached for a multivalent system. It is reminded that the maximum rate for an enzyme is obtained when all of the enzyme is saturated with substrate. For a multivalent system, the question is whether all catalytic sites can be simultaneously saturated with substrates because of geometric constraints. For example, consider a system, like the example discussed here, in which the catalytic site is composed of two neighbouring head groups. In the case where the multivalent system contains an odd number of catalytic head groups, this implies that at saturation not all head groups participate in catalysis. This leads to an intrinsic underestimation of the value for kcat. Alternatively, a non‐optimal saturation level may result from geometric constraints. Although it may be the cause for the observed lower activity of dendrimer D4 compared with D8, it is evident that this intrinsic effect is much harder to quantify. Yet, its importance emerges in a clear manner from the analysis of catalytic SAMs (see Section 7.5.2).
Even after normalization of the kcat values, a (now linear) increase of kcat/KM is observed as a function of valency driven by a decrease in KM. Although less strong, this still points to a positive dendritic effect. However, the KM value must also be interpreted with caution because of the multivalent nature of the catalyst. This is exemplified with a simple case in which catalysis by a dimeric and a tetrameric catalyst is compared (Figure 7.11). As before, catalysis requires the cooperative interaction of two head groups. In the case for example where 12 head groups are clustered in dimers, a total number of 6 binding sites are present in the catalyst. However, the same number of head groups can create 18 potential binding sites in the tetrameric catalyst. This points to an important aspect of multivalent catalysts: clustering of catalytic units in a multivalent system leads to a significant increase in the apparent number of binding sites. The consequence is an increase in the apparent affinity of the substrate for the multivalent catalysts, leading towards an apparent decrease in KM upon fitting the saturation profile to Equation 7.1. Simulations using a theoretical model indeed show that the apparent affinity of the substrate increases as a function of the valency of the system (with the same microscopic binding constant KM for each site). This leads to the important conclusion that the (generally) observed increase in binding affinity as a function of valency is an intrinsic phenomenon of multivalent catalysts and by itself does not necessarily require a chemical explanation. So, how to interpret this effect? Evidently, it affects only KM and not kcat, because at saturation the same number of substrate molecules can be accommodated by the multivalent catalyst, independent of whether these are dimeric or tetrameric. Basically, it tells us that a multivalent catalyst is intrinsically able to operate more efficiently at lower substrate concentrations. An important design principle for multivalent catalysts is thus the efficacy in creating catalytic sites.

Figure 7.11 Effect of the clustering of catalytic units on the creation of catalytic sites in multivalent models containing two or four catalytic units. Each catalytic unit is depicted by a black circle and each catalytic site by a line connecting two catalytic units.
Source: Ref. [32]. Reproduced with permission of American Chemical Society.
7.5.2 Catalytic 3D SAMs on Au NPs
In a follow‐up study, we exploited the same principle to study the catalytic efficacy of SAMs on Au NPs composed of mixtures of a catalytic thiol (containing the identical TACN · Zn2+ head group) and an inert thiol (containing a triethylene glycol head group) (Figure 7.12a) [33]. A fundamental question regarding mixed monolayer protected Au NPs regards the spatial distribution of the different thiols on the Au surface, which can be either random or phase‐separated in domains [34,35]. Model simulations analogous to those described before indicated that a different behaviour of the ‘overall’ Michaelis–Menten parameters as a function of surface thiol ratio would be observed for both situations (Figure 7.12b) [33]. In the case where domains are formed, the model predicts that both KM and kcat are nearly independent of the mole fraction of catalytic thiols in the monolayer. This is caused by the fact that the number of potential binding sites and the saturation level of the multivalent catalyst are identical for a monolayer composed of just catalytic head groups and a mixed monolayer in which these are clustered together, obviously after normalization for the total number of catalytic groups present. On the other hand, a statistical random distribution implies that as the mole fractions of catalytic groups decreases (starting from 0.3 to 0.4), the number of groups present in small clusters or even as isolated units increases. Consequently, the efficacy of the multivalent catalyst in generating binding sites for the substrate diminishes (lower apparent binding affinity, thus observed increase in KM) and the saturation level of the multivalent catalyst is also reduced (lower kcat). This was indeed experimentally observed when the catalytic activity of a series of NPs with different ratios of catalytic and inert thiols was studied. This led to the conclusion that these were distributed in a random fashion in the monolayer. This was later confirmed by an alternative study in which the binding affinity of fluorescent oligoanions to the mixed monolayer was studied as a function of the monolayer composition [36].
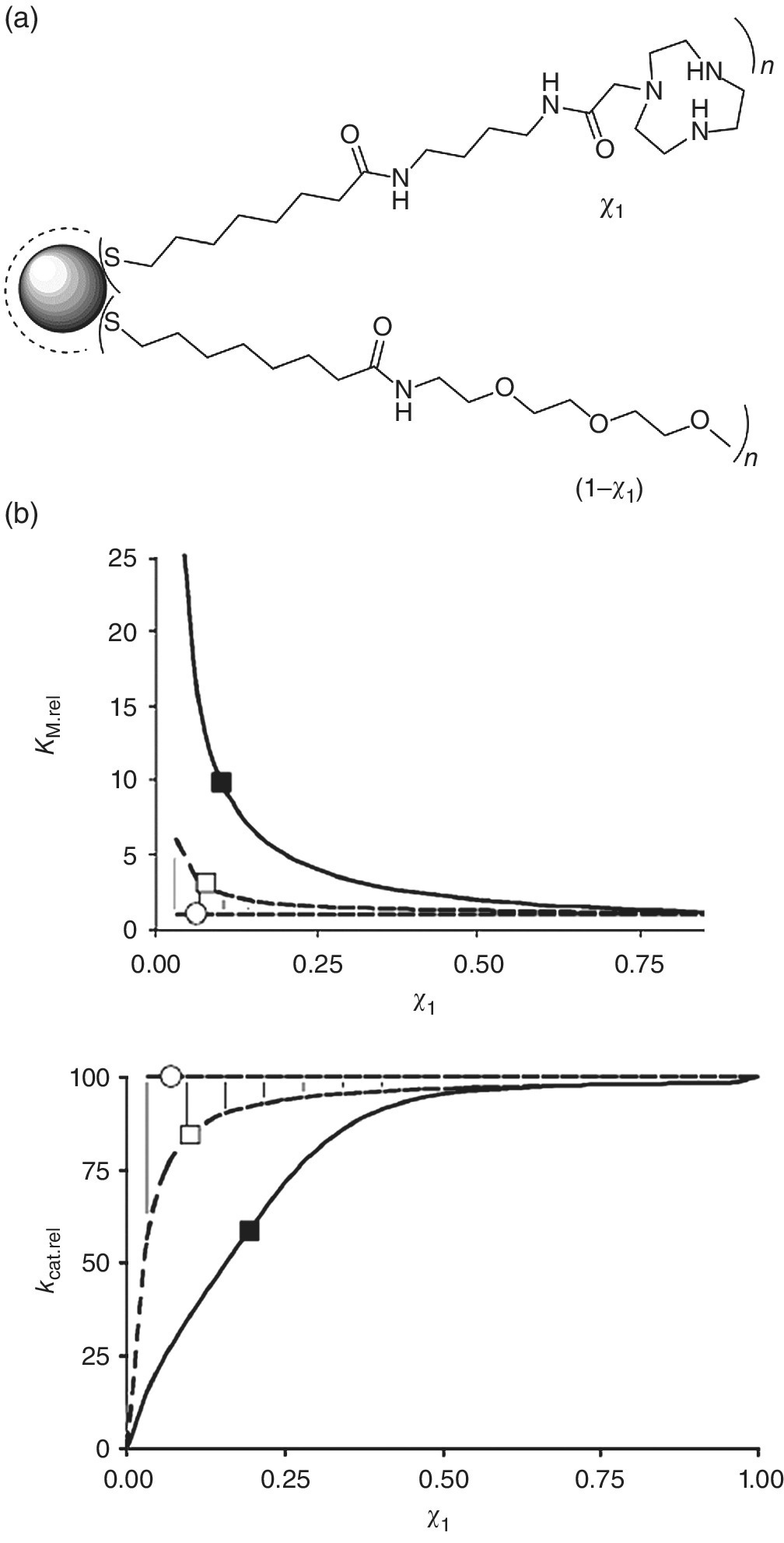
Figure 7.12 (a) Au NPs functionalized with catalytic and inert thiols. (b) Simulated correlations between KM and kcat as a function of the mole fraction χ1. The solid line marked with ( ) represents the correlation in the case where the catalytic thiols are randomly distributed in the monolayer. The dashed line marked with (
) represents the correlation in the case where the catalytic thiols are randomly distributed in the monolayer. The dashed line marked with ( ) represents homodomain formation of the catalytic thiol after monolayer formation. The dashed line marked with (
) represents homodomain formation of the catalytic thiol after monolayer formation. The dashed line marked with ( ) represents the self‐sorting of thiols on different NPs during monolayer formation (extreme case of phase separation).
) represents the self‐sorting of thiols on different NPs during monolayer formation (extreme case of phase separation).
Source: Ref. [33]. Reproduced with permission of John Wiley and Sons.
7.6 Multivalent Catalysts and Multivalent Substrates
The discussion so far has been focused on the use of multivalent structures to create catalytic sites or different chemical environments. An entirely different aspect of multivalent catalysts comes into play when these interact with multivalent substrates. This is the closest analogue compared with the multivalent binding interactions between two partners discussed in most of the other chapters in this book. Yet, related to catalysis it is an argument that has received relatively little attention [37,38]. Recently, a first attempt was made by McKay and Finn [39] to qualitatively describe what happens when both the catalyst and substrate are attached to a multivalent dendritic support. The reaction under investigation is the hydrazone formation between the aldehyde groups attached to the periphery of PAMAM‐dendrimers of different generation (with 16, 31 and 61 end groups, respectively) and nitrobenzoxadiazole hydrazine (Figure 7.13). This reaction is catalysed by anthranilic acid which forms a Schiff base with the aldehyde as intermediate. Also this catalyst was supported on PAMAM‐dendrimers of the same generations. The reaction rates could be easily determined from UV‐visible measurements. Compared with the uncatalysed background rate of the two monovalent substrates, it was found that the monovalent catalyst caused a modest three‐fold increase in activity. Incorporation of the substrate in the dendrimer caused a slight increase in background reactivity, but did not affect the three‐fold increase in rate acceleration induced by the monovalent catalyst. In contrast, dendrimer‐supported catalysts caused much faster reaction rates with polyvalent substrates, but not with monovalent ones. Depending on the valency of both systems, rate accelerations ranging from 90 to 1300 were measured relative to the strictly monovalent reference catalyst. The authors suggest two reasons that may be at the origin of this effect. The first is an enhanced initial binding of the substrates to the catalysts favoured by a high local concentration (which favours the re‐association step). The second reason is that after having catalysed the formation of the hydrazone product, re‐connection of the catalyst to a neighbouring substrate is favoured because of the high local concentration. It may also well be that a neighbouring catalyst already activates a second substrate while the first catalytic unit is still attached to the multivalent substrate. This is referred to as enhanced processivity, or a ‘rolling’ mechanism, which transmits the image of a catalyst rolling over the multivalent substrate surface. Strong support for these hypotheses came from a study of multivalent substrates and catalysts containing a lower mole fraction of active species on the dendrimer surface. A significant drop in reactivity for both multivalent systems was observed when the functional group density on the scaffold dropped from 75 to 50%. However, the simultaneous variation of both substrate and catalyst loadings produced a dramatic drop when the functional group density was reduced to 75%.

Figure 7.13 (a) Intermediates in the nucleophilic catalysis of hydrazone formation by monovalent or polyvalent anthranilic acid. The equilibrium on the bottom left shows the concomitant anthranilate‐catalysed hydrolysis of the hydrazone that establishes Keq. (b) Overall catalytic scheme. C, catalyst; S, substrate; R, capturing reagent, such as nitrobenzoxadiazole hydrazine in the present case; P, product; k1, k‐1, initial association/dissociation rate constant, respectively; k3, product‐forming step; k‐3, catalysed product‐decomposition step; k‐4, escape ~ k‐1; k5, polyvalent association rate constant (‘rolling’ rate). Letter designations (k3a, k3b, etc.) denote the rates of the same fundamental step; these rates may differ in different cycles, probably only slightly in the early stages of the reaction. Accelerated substrate transformation may occur if ‘rolling’ (k5) is much faster than diffusion‐controlled steps (k‐4, k1).
Source: Ref. [39]. Reproduced with permission of John Wiley and Sons.
A practical application of the combination of multivalent catalysts and substrates was provided by Bonomi et al. [40]. They immobilized BAPA•Zn(II) complexes on Au NPs together with triethylene glycol‐terminated thiols (Figure 7.14) [40]. The obtained NP showed an extraordinary activity in the cleavage of the DNA model substrate bis‐p‐nitrophenyl phosphate (BNP) with a rate acceleration of over 5 orders of magnitude over the background. Importantly, comparison with the monomeric complex BAPA•Zn(II) showed that insertion in the SAM caused a 100‐fold gain in reactivity. Similar as observed for the NPs discussed above (Figure 7.5), the major source for this rate acceleration originates from the formation of dinuclear catalytic sites in the SAM. Nonetheless, the most intriguing aspect of this NP‐based catalyst appeared when DNA was used as a substrate. Incubation of pBR 322 plasmid DNA with the catalyst resulted in a significant phosphodiester cleavage. Under the same conditions, the monomeric catalyst showed no activity at all. Remarkably, the amount of linear DNA formed was 50% larger than that of nicked DNA, the formation of which is statistically much more favourable. This result indicates that the multivalent NP‐based catalyst preferably performs double strand cleavage. This seems to be a consequence of the multivalency of the NPs, which generates multiple contacts between catalyst and cleavable bonds upon formation of the catalyst–substrate complex.

Figure 7.14 Catalytic Au NPs for the cleavage of phosphodiesters. The result of the cleavage of plasmid DNA and a cartoon representing the interaction between the catalyst and the biopolymer are also shown.
Source: Ref. [40]. Reproduced with permission of American Chemical Society.
7.7 Conclusions
As discussed in Section 7.1, multivalent catalysts are most frequently associated with numerous homogeneous catalysts attached to a multivalent scaffold, prepared with the scope to facilitate recovery and re‐usage of the precious catalysts. A good design implied that no loss of catalytic performance was observed compared with the reference homogeneous catalyst and/or during subsequent cycles. Occasional improvements of the catalytic performance (e.g. in rate or enantioselectivity) was a welcome additional bonus not sought for. Yet, the examples discussed in this chapter illustrate that the potential of multivalent catalysts goes far beyond this. Design principles are emerging aimed at exploiting the ability of multivalent systems to induce cooperativity between functional groups. At difference with traditional multivalent catalysts, this implies that multivalency becomes a prerequisite for observing catalytic activity. The possibility to alter in a controlled manner the local chemical environment in a multivalent catalyst becomes an attractive strategy to control actively the catalytic performance. It is further shown that multivalent catalysts bear intrinsic advantages compared with monovalent ones when substrate binding to the catalytic system plays a role. The enhanced number of potential binding sites in a multivalent catalyst makes it perform better (in terms of rate) at low concentration compared with the monomeric reference. Overall, the examples discussed here mark the importance of multivalency as a design principle for catalysts. The next challenge is to develop methodology aimed at maximizing cooperativity in these catalysts, create local enzyme‐like pockets of which the (chiral) chemical environment can be controlled, and widen the reaction scope, which is currently still rather limited.
Acknowledgements
Funding from the European Union Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No. 642793 is acknowledged.
References
- 1 D. Mery, D. Astruc, Dendritic catalysis: Major concepts and recent progress, Coord. Chem. Rev. 2006, 250, 1965–1979.
- 2 S. Roy, M. A. Pericas, Functionalized nanoparticles as catalysts for enantioselective processes, Org. Biomol. Chem. 2009, 7, 2669–2677.
- 3 J. M. Fraile, J. I. Garcia, J. A. Mayoral, Noncovalent immobilization of enantioselective catalysts, Chem. Rev. 2009, 109, 360–417.
- 4 A. Schatz, O. Reiser, W. J. Stark, Nanoparticles as semi‐heterogeneous catalyst supports, Chem. Eur. J. 2010, 16, 8950–8967.
- 5 D. Astruc, E. Boisselier, C. Ornelas, Dendrimers designed for functions: From physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine, Chem. Rev. 2010, 110, 1857–1959.
- 6 A. Thomas, M. Driess, Bridging the materials gap in catalysis: Entrapment of molecular catalysts in functional supports and beyond, Angew. Chem. Int. Ed. 2009, 48, 1890–1892.
- 7 S. Javor, E. Delort, T. Darbre, J. L. Reymond, A peptide dendrimer enzyme model with a single catalytic site at the core, J. Am. Chem. Soc. 2007, 129, 13238–13246.
- 8 T. Darbre, J. L. Reymond, Peptide dendrimers as artificial enzymes, receptors, and drug‐delivery agents, Acc. Chem. Res. 2006, 39, 925–934.
- 9 J. Kofoed, J. L. Reymond, Dendrimers as artificial enzymes, Curr. Opin. Chem. Biol. 2005, 9, 656–664.
- 10 L. Pasquato, P. Pengo, P. Scrimin, Nanozymes: Functional nanoparticle‐based catalysts, Supramol. Chem. 2005, 17, 163–171.
- 11 G. Pieters, L. J. Prins, Catalytic self‐assembled monolayers on gold nanoparticles, New J. Chem. 2012, 36, 1931–1939.
- 12 T. Belser, M. Stohr, A. Pfaltz, Immobilization of rhodium complexes at thiolate monolayers on gold surfaces: Catalytic and structural studies, J. Am. Chem. Soc. 2005, 127, 8720–8731.
- 13 C. C. Paluti, E. S. Gawalt, Immobilized aza‐bis(oxazoline) copper catalysts on SAMs: Selectivity dependence on catalytic site embedding, J. Cat. 2009, 267, 105–113.
- 14 C. C. Paluti, E. S. Gawalt, Immobilized aza‐bis(oxazoline) copper catalysts on alkanethiol self‐assembled monolayers on gold: Selectivity dependence on surface electronic environments, J. Cat. 2010, 275, 149–157.
- 15 C. Guarise, F. Manea, G. Zaupa, L. Pasquato, L. J. Prins, P. Scrimin, Cooperative nanosystems, J. Pept. Sci. 2008, 14, 174–183.
- 16 E. Delort, T. Darbre, J. L. Reymond, A strong positive dendritic effect in a peptide dendrimer‐catalyzed ester hydrolysis reaction, J. Am. Chem. Soc. 2004, 126, 15642–15643.
- 17 L. Ropartz, R. E. Morris, D. F. Foster, D. J. Cole‐Hamilton, Increased selectivity in hydroformylation reactions using dendrimer based catalysts; a positive dendrimer effect, Chem. Commun. 2001, 361–362.
- 18 A. Dahan, M. Portnoy, Remarkable dendritic effect in the polymer‐supported catalysis of the Heck arylation of olefins, Org. Lett. 2003, 5, 1197–1200.
- 19 Y. Ribourdouille, G. D. Engel, M. Richard‐Plouet, L. H. Gade, A strongly positive dendrimer effect in asymmetric catalysis: Allylic aminations with Pyrphos‐palladium functionalised PPI and PAMAM dendrimers, Chem. Commun. 2003, 1228–1229.
- 20 B. Helms, J. M. J. Frechet, The dendrimer effect in homogeneous catalysis, Adv. Synth. Cat. 2006, 348, 1125–1148.
- 21 D. E. Wilcox, Binuclear metallohydrolases, Chem. Rev. 1996, 96, 2435–2458.
- 22 F. Mancin, P. Scrimin, P. Tecilla, U. Tonellato, Artificial metallonucleases, Chem. Commun. 2005, 2540–2548.
- 23 F. Manea, F. B. Houillon, L. Pasquato, P. Scrimin, Nanozymes: Gold‐nanoparticle‐based transphosphorylation catalysts, Angew. Chem. Int. Ed. 2004, 43, 6165–6169.
- 24 K. B. Hansen, J. L. Leighton, E. N. Jacobsen, On the mechanism of asymmetric nucleophilic ring‐opening of epoxides catalyzed by (salen)CrIII complexes, J. Am. Chem. Soc. 1996, 118, 10924–10925.
- 25 L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond, E. N. Jacobsen, Mechanistic investigation leads to a synthetic improvement in the hydrolytic kinetic resolution of terminal epoxides, J. Am. Chem. Soc. 2004, 126, 1360–1362.
- 26 R. Breinbauer, E. N. Jacobsen, Cooperative asymmetric catalysis with dendirmeric Co(salen) complexes, Angew. Chem. Int. Ed. 2000, 39, 3604–3607.
- 27 T. Belser, E. N. Jacobsen, Cooperative catalysis in the hydrolytic kinetic resolution of epoxides by chiral (salen)Co(III) complexes immobilized on gold colloids, Adv. Synth. Cat. 2008, 350, 967–971.
- 28 P. Pengo, S. Polizzi, L. Pasquato, P. Scrimin, Carboxylate – Imidazole cooperativity in dipeptide‐functionalized gold nanoparticles with esterase‐like activity, J. Am. Chem. Soc. 2005, 127, 1616–1617.
- 29 D. Zaramella, P. Scrimin, L. J. Prins, Self‐Assembly of a catalytic multivalent peptide‐nanoparticle complex, J. Am. Chem. Soc. 2012, 134, 8396–8399.
- 30 D. Zaramella, P. Scrimin, L. J. Prins, Catalysis of transesterification reactions by a self‐assembled nanosystem, Int. J. Mol. Sci. 2013, 14, 2011–2021.
- 31 H. Wei, E. K. Wang, Nanomaterials with enzyme‐like characteristics (nanozymes): next‐generation artificial enzymes, Chem. Soc. Rev. 2013, 42, 6060–6093.
- 32 G. Zaupa, P. Scrimin, L. J. Prins, Origin of the dendritic effect in multivalent enzyme‐like catalysts, J. Am. Chem. Soc. 2008, 130, 5699–5709.
- 33 G. Zaupa, C. Mora, R. Bonomi, L. J. Prins, P. Scrimin, Catalytic self‐assembled monolayers on Au nanoparticles: The source of catalysis of a transphosphorylation reaction, Chem. Eur. J. 2011, 17, 4879–4889.
- 34 C. Gentilini, L. Pasquato, Morphology of mixed‐monolayers protecting metal nanoparticles, J. Mater. Chem. 2010, 20, 1403–1412.
- 35 D. Rodriguez‐Fernandez, L. M. Liz‐Marzan, Metallic Janus and patchy particles, Part. Part. Syst. Charact. 2013, 30, 46–60.
- 36 R. Bonomi, A. Cazzolaro, L. J. Prins, Assessment of the morphology of mixed SAMs on Au nanoparticles using a fluorescent probe, Chem. Commun. 2011, 47, 445–447.
- 37 X. L. Liao, R. T. Petty, M. Mrksich, A spatially propagating biochemical reaction, Angew. Chem. Int. Ed. 2011, 50, 706–708.
- 38 R. A. Pavlick, S. Sengupta, T. McFadden, H. Zhang, A. Sen, A polymerization‐powered motor, Angew. Chem. Int. Ed. 2011, 50, 9374–9377.
- 39 C. S. McKay, M. G. Finn, Polyvalent catalysts operating on polyvalent substrates: A model for surface‐controlled reactivity, Angew. Chem. Int. Ed. 2016, DOI: 10.1002/anie.201602797.
- 40 R. Bonomi, F. Selvestrel, V. Lombardo, C. Sissi, S. Polizzi, F. Mancin, U. Tonellato, P. Scrimin, Phosphate diester and DNA hydrolysis by a multivalent, nanoparticle‐based catalyst, J. Am. Chem. Soc. 2008, 130, 15744.