
Chapter 26
PARASITES
William N. Yang
CRYPTOSPORIDIUM PARVUM
Common names for disease: Cryptosporidiosis
Many species of Cryptosporidium exist that infect humans and a wide range of animals. Although Cryptosporidium parvum, found in young cattle and other herbivores, and Cryptosporidium hominis, the human-adapted species, (formerly known as C. parvum anthroponotic genotype or genotype 1) are the most prevalent species causing disease in humans, infections by Cryptosporidium felis, Cryptosporidium meleagridis, Cryptosporidium canis, and Cryptosporidium muris have also been reported.1
Occupational setting
Farmers, animal handlers, veterinarians, laboratory personnel, healthcare workers, daycare workers, and divers are at risk from exposure.1–4
Exposure (route)
Human cryptosporidiosis was first reported in 1976. Its characteristics are having a very low infectious dose (10–1000 oocysts), being severe in immunocompromised populations, being very resistant to antiparasitic therapy, and having human and nonhuman reservoirs. Cryptosporidiosis is most common in developing countries, with waterborne, foodborne, person-to-person, and zoonotic transmission pathways. It still remains a significant pathogen of children and the elderly in industrialized nations and can cause major outbreaks when public health measures break down. It is a common cause of wasting and lethal diarrhea in HIV-infected persons, and acute and persistent disease among farmers, animal handlers, and veterinarians results most commonly from exposure to infected calves and other farm animals. The prevalence of infection is higher in young animals, such as calves and lambs.5 An outbreak of cryptosporidiosis occurred among firefighters responding to a fire involving a barn that contained preweaned calves.6 Other sources of infection are contaminated water supplies, canals, lakes or rivers, and swimming and wading pools, which have been contaminated by animal or human sewage. Occupational and recreational divers are at risk. Person-to-person (fecal–oral) infection may also occur in healthcare and daycare settings.4,7,8 The 1993 cryptosporidiosis waterborne outbreak in Milwaukee, Wisconsin, was the largest waterborne outbreak in US history, affecting over 400 000 people. The outbreak was estimated to cost over 96 million dollars in lost productivity and medical costs.9
Pathobiology
Cryptosporidium parvum is an intracellular but extracytoplasmic protozoan parasite. When a human ingests the oocyst that has been passed in the feces of an infected host, the oocyst wall dissolves, and sporozoite forms invade the host gastrointestinal (GI) epithelial cells. They pass through a trophozoite stage and an asexual multiplication stage that results in schizonts. These schizonts can reinvade the host or continue to a sexual multiplication stage that leads to new oocysts that are passed outside the body. The sporulated oocyst is the only developmental stage that occurs extracellularly. Since this part of the cycle is completed before the oocyst is excreted, the oocysts are immediately infectious when passed in feces.5In immunocompromised persons, the ability to reinfect the host can lead to continuing infection (Figure 26.1).

FIGURE 26.1 Cryptosporidium Life Cycle. Sporulated oocysts, containing 4 sporozoites, are excreted by the infected host through feces and possibly other routes such as respiratory secretions  . Transmission of Cryptosporidium parvum and C. hominis occurs mainly through contact with contaminated water (e.g., drinking or recreational water). Occasionally food sources, such as chicken salad, may serve as vehicles for transmission. Many outbreaks in the United States have occurred in waterparks, community swimming pools, and day care centers. Zoonotic and anthroponotic transmission of C. parvum and anthroponotic transmission of C. hominis occur through exposure to infected animals or exposure to water contaminated by feces of infected animals
. Transmission of Cryptosporidium parvum and C. hominis occurs mainly through contact with contaminated water (e.g., drinking or recreational water). Occasionally food sources, such as chicken salad, may serve as vehicles for transmission. Many outbreaks in the United States have occurred in waterparks, community swimming pools, and day care centers. Zoonotic and anthroponotic transmission of C. parvum and anthroponotic transmission of C. hominis occur through exposure to infected animals or exposure to water contaminated by feces of infected animals  . Following ingestion (and possibly inhalation) by a suitable host
. Following ingestion (and possibly inhalation) by a suitable host  , excystation
, excystation  occurs. The sporozoites are released and parasitize epithelial cells (
occurs. The sporozoites are released and parasitize epithelial cells ( ,
,  ) of the gastrointestinal tract or other tissues such as the respiratory tract. In these cells, the parasites undergo asexual multiplication (schizogony or merogony) (
) of the gastrointestinal tract or other tissues such as the respiratory tract. In these cells, the parasites undergo asexual multiplication (schizogony or merogony) ( ,
,  ,
,  ) and then sexual multiplication (gametogony) producing microgamonts (male)
) and then sexual multiplication (gametogony) producing microgamonts (male)  and macrogamonts (female)
and macrogamonts (female)  . Upon fertilization of the macrogamonts by the microgametes (
. Upon fertilization of the macrogamonts by the microgametes ( ), oocysts (
), oocysts ( ,
,  ) develop that sporulate in the infected host. Two different types of oocysts are produced, the thick-walled, which is commonly excreted from the host , and the thin-walled oocyst , which is primarily involved in autoinfection. Oocysts are infective upon excretion, thus permitting direct and immediate fecal-oral transmission. Centers for Disease Control and Prevention. Parasites – Cryptosporidium.
) develop that sporulate in the infected host. Two different types of oocysts are produced, the thick-walled, which is commonly excreted from the host , and the thin-walled oocyst , which is primarily involved in autoinfection. Oocysts are infective upon excretion, thus permitting direct and immediate fecal-oral transmission. Centers for Disease Control and Prevention. Parasites – Cryptosporidium.
Cryptosporidium hominis and C. parvum are the cause of about 75 and 20% of human disease.
Cryptosporidium hominis is more virulent in humans than other species and only infects humans.
Cryptosporidium parvum infects cattle and sheep. A third common human pathogen is C. meleagridis, an avian species.1 In developing countries ~20% of children with diarrhea have cryptosporidiosis compared with 1–5% in North America and Europe. Cryptosporidiosis prevalence is higher in rural areas. Malnutrition, HIV/AIDS, and other immunocompromising conditions significantly increase the risk, severity, and persistence of cryptosporidiosis. It is an AIDS opportunistic infection and often afflicts malnourished children with persistent diarrhea. Cryptosporidiosis-affected children may have impaired growth and cognitive function and have multiple symptomatic episodes before they acquire partial protective immunity. While Cryptosporidium parasites infect the colonic mucosa and, less commonly, the small intestine and stomach, replicating parasites can infect the entire gut from the oropharynx to the anus.
The incubation period ranges from 4 to 10 days after oocyst ingestion. Excretion of infectious oocysts in feces begins when symptoms begin and can persist for months. Oocysts of Cryptosporidium spp. are immediately infectious when they are passed in stool. Internal autoinfection is common and may be the reason for the persistent nature of the disease in immunocompromised people. The immune status of the person determines the severity and length of the symptoms. In immunocompetent persons, the symptoms begin quickly, after a 2–12-day incubation period, and then last from 7 to 14 days with a mean of 9–11 days.1,5
Symptoms of disease include watery diarrhea (the most frequent symptom), cramping abdominal pain, weight loss, and flatulence. Less common symptoms are nausea, vomiting, myalgias, and fever. While the small intestine is the site most commonly affected, symptomatic Cryptosporidium infections have also been found in other organs including other digestive tract organs, the lungs, and possibly conjunctiva. 1,5
In immunocompetent persons, symptoms are usually self-limited (1–2 weeks). In immunocompromised patients, especially those with CD4 counts <200/μL, symptoms can be chronic and more severe. The diarrhea can last for months and lead to dehydration, electrolyte imbalance, and malnutrition and can be life threatening.1 While cryptosporidial cysts can be found in the stool, blood and white blood cells are usually absent. Oocysts may remain in the stool for 8–50 days (mean 12–14 days) after the clinical symptoms have ended.
AIDS patients with Cryptosporidium sclerosing cholangitis and biliary involvement usually have elevated serum alkaline phosphatase levels and right upper quadrant pain. Symptoms in patients with reversible causes of immunodeficiencies usually resolve quickly when the cause of the immunosuppression is eliminated.1,5
Diagnosis
Cryptosporidiosis is usually mild and underdiagnosed since often no diagnostic testing is done. Suspicion for Cryptosporidium as the cause of persistent or chronic diarrhea is increased if the individual is malnourished, has an immunosuppressive condition, or has another risk factor. The incubation period is longer (~1 week) than it is for viral or bacterial diseases (1–3 days) yet shorter than for giardiasis (~2 weeks). Giardia and Isospora infections may also mimic cryptosporidiosis.1
The diagnosis of cryptosporidiosis can be made by microscopic examination of stool specimens or by examination of biopsied tissue from infected intestine, biliary system, or respiratory tract. With a modified acid-fast technique, the oocysts are red-stained and can be differentiated from similar-appearing yeast forms that do not take up the acid-fast stain. Strict morphologic criteria must be applied to the diagnosis to avoid confusion with other oocysts, such as Cyclospora oocysts, which are much larger (10 mm).10 Antigen detection by ELISA, PCR, or a direct fluorescent antibody (DFA) assay are also available and have the advantage of high sensitivity.1,5
Treatment
In the immunocompetent host, the infection is self-limited. Oral or intravenous rehydration, antimotility agents, and nutritional support are used for the symptomatic treatment of diarrhea. Nitazoxanide (NTZ) is the only Food and Drug Administration (FDA)-approved drug for cryptosporidiosis. NTZ can shorten clinical disease by a day or so on average and reduce parasite loads in immunocompetent patients, but it is not consistently effective for immunodeficient persons. Those on corticosteroids or cytotoxic drugs will recover if the agents are stopped.1,5 Since highly active antiretroviral therapy (HAART) restores cell-mediated immunity and has been shown to decrease the prevalence of cryptosporidiosis in HIV-infected patients, it is considered the most effective treatment and prophylaxis for AIDS patients.1,5
Medical surveillance
Occupationally exposed or at-risk workers should be monitored for signs or symptoms of diarrhea. In conjunction with medical surveillance, workers should receive education about the sources of exposure and preventive measures.
Prevention
Prevention is based on practicing good hygiene and educating staff about the way cryptosporidiosis is spread in childcare or animal settings. Good hygiene includes good hand washing before preparing or eating food, after toilet use, in daycare settings after changing diapers or cleaning up a child who has used the toilet, and in animal settings after handling animals or animal waste. Good hand washing is defined as washing hands with soap and water for at least 20 seconds, rubbing hands together vigorously, and scrubbing all surfaces.5
In farm and institutional settings, personal hygiene practices, appropriate use of disposable gloves, and environmental sanitation should be emphasized. Strict enteric precautions should be practiced around infected animals or humans. Eliminating the disease in farm animals decreases the risk for farmers and veterinary personnel.1
Cooking kills Cryptosporidium in food, but vegetables and fruits to be consumed uncooked must be thoroughly washed with potable water before eating. Oocysts are killed by pasteurization (72°C for 5 seconds), heating to 60°C for 30 minutes, or freezing at −7°C for 1 hour. No safe and effective disinfectant has been identified for decontaminating produce.1
Cryptosporidium is extremely tolerant to chlorine, and chlorine-based disinfectants (e.g., bleach solutions) will not kill Cryptosporidium.1 Drinking water sources must be protected from human and animal fecal material from surface runoff. Maintaining proper filtration procedures for drinking water supplies will minimize infection but may not prevent it due to the small size of the oocysts.10 Travelers should minimize their exposure by avoiding untreated water and uncooked fruits and vegetables. The oocyst will survive in 3% hypochlorite solution, iodophors, benzalkonium chloride, and 5% formaldehyde. Reverse osmosis filters can remove all oocysts but require low-turbidity source water to prevent plugging. Contaminated water can be purified for personal use by heating the water to a rolling boil for 1 minute or using a filter that has been tested and rated by the National Sanitation Foundation (NSF) Standard 53 or NSF Standard 58 for cyst and oocyst reduction; filtered water will need additional treatment to kill or inactivate bacteria and viruses.5
Cryptosporidiosis can be prevented in the immunocompromised by education and by drinking only filtered or boiled water, ingesting only cooked food, thorough hand washing, avoiding bathing or swimming in water used by other people or animals, and avoiding contact with people or animals (especially calves and lambs) with diarrhea. Companion dogs and cats should be examined and cleared of infection by a veterinarian.1
Immunocompromised persons should not have contact with any animal that has diarrhea. They should also realize that there is risk of infection from the accidental ingestion of recreational lake, river, or swimming pool water due to intermittent contamination with cryptosporidia from human or animal waste.1,5
References
- 1. Xiao L, Griffiths JK. Cryptosporidiosis. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases, 9th edn. London/New York: Saunders/Elsevier, 2013:673–80.
- 2. Current VVL, Reese NC, Ernst JY, et al. Human cryptosporidiosis in immunocompetent and immunodeficient persons. Studies of an outbreak and experimental transmission. N Engl J Med 1983; 308:1252–7.
- 3. Pohjola S, Oksanen H, Jokipii L, et al. Outbreak of cryptosporidiosis among veterinary students. Scand J Infect Dis 1986; 18:173–8.
- 4. Artieda J, Basterrechea M, Arriola L, et al. Outbreak of cryptosporidiosis in a child day-care centre in Gipuzkoa, Spain, October to December 2011. Euro Surveill 2012; 17(5):pii: 20070.
- 5. Centers for Disease Control and Prevention. Parasites – Cryptosporidium. http://www.cdc.gov/parasites/crypto/ (accessed October 15, 2014).
- 6. Centers for Disease Control and Prevention. Outbreak of cryptosporidiosis associated with a firefighting response — Indiana and Michigan, June 2011. MMWR 2012; 61(9):153–6.
- 7. Koch KL, Phillips DJ, Aber RC, et al. Cryptosporidiosis in hospital personnel. Evidence for person-to-person transmission. Ann Intern Med 1985; 102(5):593–6.
- 8. Xiao G, Wang Z, Chen J, et al. Occurrence and infection risk of waterborne pathogens in Wanzhou watershed of the Three Gorges Reservoir, China. J Environ Sci (China) 2013; 25(9):1913–24.
- 9. Corso PS, Kramer MH, Blair KA, et al. Cost of illness in the 1993 waterborne Cryptosporidium outbreak, Milwaukee, Wisconsin. Emerg Infect Dis 2003; 9(4):426–31.
- 10. MacKenzie WR, Hoxie NJ, Proctor ME, et al. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N Engl J Med 1994; 331:161–7.
CYCLOSPORIASIS
Common names for disease: None
Cyclosporiasis is an intestinal infection with the coccidian organism Cyclospora cayetanensis. Symptoms of cyclosporiasis begin an average of 7 days (range 2 days to ≥2 weeks) after ingestion of sporulated cysts—the infective form of the parasite.
Symptoms can include watery diarrhea, fatigue, anorexia, nausea, cramping, increased gas, and bloating. If a person ill with cyclosporiasis is not treated, symptoms can persist for several weeks to a month or more. Some symptoms, such as diarrhea, can return, and other symptoms, such as muscle aches and fatigue, may continue after the gastrointestinal symptoms have gone away. The infection usually is not life threatening.
The organism was definitively identified in 1993, after being independently discovered by four different researchers dating back to 1979. Before 1996, cyclosporiasis in North America was associated with overseas travelers. In 1996, a large outbreak in North America occurred in the spring and summer and was associated with eating Guatemalan raspberries.1–3 Cyclosporiasis is most common in tropical and subtropical regions. While the risk for infection is seasonal, there is no consistent pattern regarding the time of year or the environmental conditions, such as temperature or rainfall. In the United States foodborne outbreaks of cyclosporiasis since the mid-1990s have been linked to various types of imported fresh produce including raspberries, basil, snow peas, and mesclun lettuce. No commercially frozen or canned produce has been implicated.1
Even though here have not been any large outbreaks of cyclosporiasis recently, in 2013 the Centers for Disease Control and Prevention (CDC) reported a total of 631 persons infected with C. cayetanensis from 25 states and New York City. Most illness occurred between mid-June and mid-July with eight percent of ill persons hospitalized and no deaths reported. Ill persons ranged from less than 1 year to 94 years, with a median age of 52 years. 1
Occupational setting
Cyclosporiasis can be an important cause of diarrhea domestically and internationally as a result of contaminated food or water. Cyclospora has been found throughout the world in both immunocompetent and immunosuppressed persons. Cyclospora infected 7% of the Nepal American Embassy community during the 1992 Cyclospora season and was the cause of 11% of the cases of diarrhea seen at the Canadian International Water and Energy Consultants Clinic in Kathmandu. Cyclospora cayetanensis should be considered as a possible cause of diarrhea in an individual with prolonged watery diarrhea with a history of travel to a developing country.4
Cyclospora has also been linked to an outbreak of foodborne illness on a passenger ship.5 Epidemiology studies done in Guatemala have found Cyclospora in farm workers in one study but not in another. With the globalization of food systems, a large part of the US food supply now comes from areas where Cyclospora is endemic and where methods for inactivation and complete removal of Cyclospora oocysts from contaminated produce have not been developed. Countries where crops are grown for local consumption and exportation need to practice good agricultural methods and use filtered or otherwise decontaminated irrigation water. Worldwide food safety education and training is important to insure safe food and water for all.6
Exposure (route)
While the transmission of Cyclospora appears to be fecal–oral, there is no definite evidence of person-to-person spread. Waterborne transmission and foodborne transmission evidence does exist. A water supply from a hospital building at a Chicago hospital was implicated as the source of a 1990 summer outbreak in the hospital staff, and water was associated with a case–control study of cyclosporiasis in Kathmandu.5 The large outbreak that occurred in 1996 was related to the consumption of raspberries imported from Guatemala.3 In 1997, several other outbreaks in Canada and the United States were also linked to raspberries from Guatemala and to basil and mesclun lettuce.1
The mode of contamination of the raspberries is unclear. However, following the 1996 outbreak, berry growers and exporters in Guatemala, with assistance from the Food and Drug Administration (FDA) and CDC, began voluntary water quality and sanitary control measures.1,4 After another outbreak in spring 1997, Guatemala began classifying the farms, and only “low-risk” farms were allowed to export to North America.4 In 1998, the FDA banned the importation of fresh raspberries into the United States, but Canada continued to import them until June 1998. Two surveys of indigenous Peruvian children under the age of 30 months found 6 and 18% with Cyclospora cysts in their feces.1,2 Of those infected, 11 and 28%, respectively, presented with diarrhea.
Of interest is the seasonality of Cyclospora. In Nepal, the organism is present from May, just before the rainy season, until October, just after the rainy season, with the peak risk in June and July. In Peru, the season extends from January to July, with a peak from April to June. In Guatemala, the main season of risk is May to September.2
Since a large part of the US food supply comes from areas where Cyclospora is endemic and methods for inactivation and complete removal of Cyclospora oocysts from contaminated produce have not been developed, countries where crops are grown for local consumption and exportation need to practice good agricultural methods and use filtered or otherwise decontaminated irrigation water.6
Pathobiology
Cyclospora cayetanensis completes its life cycle in humans. However, the oocysts shed in the feces of infected persons must mature (sporulate) outside the host, in the environment, to become infective for someone else. Therefore, direct person-to-person (fecal–oral) transmission of Cyclospora is unlikely. The process of maturation (sporulation) is thought to require days to weeks (Figure 26.2).

FIGURE 26.2 Cyclospora Life Cycle. Some of elements of this figure were created based on an illustration by Ortega et al. Cyclospora cayetanensis. In: Advances in Parasitology: opportunistic protozoa in humans. San Diego: Academic Press; 1998. p. 399–418. When freshly passed in stools, the oocyst is not infective (thus, direct fecal-oral transmission cannot occur; this differentiates Cyclospora from another important coccidian parasite, Cryptosporidium). In the environment , sporulation occurs after days or weeks at temperatures between 22°C to 32°C, resulting in division of the sporont into two sporocysts, each containing two elongate sporozoites  . Fresh produce and water can serve as vehicles for transmission
. Fresh produce and water can serve as vehicles for transmission  and the sporulated oocysts are ingested (in contaminated food or water)
and the sporulated oocysts are ingested (in contaminated food or water)  . The oocysts excyst in the gastrointestinal tract, freeing the sporozoites which invade the epithelial cells of the small intestine
. The oocysts excyst in the gastrointestinal tract, freeing the sporozoites which invade the epithelial cells of the small intestine  . Inside the cells they undergo asexual multiplication and sexual development to mature into oocysts, which will be shed in stools
. Inside the cells they undergo asexual multiplication and sexual development to mature into oocysts, which will be shed in stools  . The potential mechanisms of contamination of food and water are still under investigation.
. The potential mechanisms of contamination of food and water are still under investigation.
Life cycle image and information courtesy of DPDx. Source: http://www.cdc.gov/parasites/cyclosporiasis/biology.html
Persons of all ages living or traveling in the tropics and subtropics may be at increased risk because cyclosporiasis is endemic in some countries in these zones. In some regions, infection appears to be seasonal, but the seasonality varies in different settings and is not well understood.
Some infected persons are asymptomatic, particularly in settings where cyclosporiasis is endemic. Among symptomatic persons, the incubation period averages ~1 week (range ~2–14 or more days). Cyclospora infects the small intestine and typically causes watery diarrhea, with frequent, sometimes explosive, stools. Other common symptoms include loss of appetite, weight loss, abdominal cramping and bloating, increased flatus, nausea, and prolonged fatigue. Vomiting, body aches, low-grade fever, and other flu-like symptoms may be also be noted. If untreated, the illness may last for a few days to a month or longer and may follow a remitting–relapsing course. Although cyclosporiasis usually is not life threatening, reported complications have included malabsorption, cholecystitis, and Reiter’s syndrome—a reactive arthritis.1
If untreated, clinical manifestations can last for an average of 6–7 weeks in immunocompetent hosts, and in HIV-positive individuals, the diarrhea can last for months. The organism can be detected in stools up to 8 weeks after the onset of symptoms.2 Asymptomatic individuals who are excreting cysts have been documented, particularly in areas where it is endemic.1
Diagnosis
The main method is identifying the typical oocysts in stools after using concentration methods and then staining stool smears with a modified acid-fast stain. Concentration techniques can increase the diagnostic yield by over 40%, which is very helpful in individuals with acute and severe disease who are shedding low number of organisms.2 Fluorescence microscopy can assist in the diagnosis, since the oocysts autofluoresce. The diagnosis can also be made by observing the oocysts in wet preparation from jejunal aspirates.5 Current CDC guidelines for the confirmation of C. cayetanensis foodborne disease outbreaks require the demonstration of the organism in stools of two or more ill persons.7 Molecular diagnostic methods, such as polymerase chain reaction (PCR) analysis, can be used to look for the parasite’s DNA in the stool. The lack of DNA information of the various strains of the parasite hinders surveillance and outbreak response.
Treatment
Most people with healthy immune systems will recover without treatment. Without treatment, the illness may last for a few days to a month or longer. Symptoms may seem to go away and then return one or more times (relapse). Antidiarrheal medicine may help reduce diarrhea, but a healthcare provider should be consulted before such medicine is taken. People who are in poor health or who have weakened immune systems may be at higher risk for severe or prolonged illness.
The medical treatment for immunocompetent adults is TMP 160 mg plus SMX 800 mg (one double-strength tablet), orally, twice a day, for 7–10 days. HIV-infected patients and others with immune compromise may need longer courses of therapy. No other alternate medications have been identified for persons who are allergic to or cannot take TMP/SMX.1
Medical surveillance
CDC conducts surveillance for cyclosporiasis. Cyclosporiasis is a nationally notifiable disease and is reportable in over 35 states. Regardless of whether cyclosporiasis is reportable in a particular state, the public and clinicians need to inform their local health department about potential cases and clusters of the disease so that appropriate investigative actions can be taken to help prevent additional cases of illness.
CDC, in collaboration with public health authorities, analyzes each reported case for epidemiological evidence of linkage to other cases to facilitate rapid identification and investigation of outbreaks. There are no molecular tools available for linking C. cayetanensis cases.1
Future sequencing of the DNA of samples of the parasite that circulate in the United States and different parts of the world and analyzing the DNA of parasites collected from individual outbreak-related cases—as well as cases not known to be linked to an outbreak—will identify potential genotyping markers and develop a new DNA-based surveillance system for cyclosporiasis.1
Prevention
On the basis of the currently available information, avoiding food or water that may have been contaminated with feces is the best way to prevent cyclosporiasis. Cyclospora oocysts are killed by boiling water but not by treatment with chlorine or iodine. No vaccine for cyclosporiasis is available. Fresh fruits, especially raspberries and salads, should be thoroughly washed before eating.1
References
- 1. Centers for Disease Control and Prevention. Cyclosporiasis. http://www.cdc.gov/parasites/cyclosporiasis/ (accessed October 21, 2014).
- 2. Shlim DR, Connor BA. Cyclosporoiasis. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Disease, 9th edn. New York: Elsevier, 2013:680–3.
- 3. Herwaldt BL, Ackers M. The cyclospora working group. An outbreak in 1996 of cyclosporiasis associated with imported raspberries. N Engl J Med 1997; 336:1548–56.
- 4. Hoge CW, Shlim DR, Rajah R, et al. Epidemiology of diarrhoeal illness associated with coccidian-like organism among travelers and foreign residents in Nepal. Lancet 1993; 341:1175–9.
- 5. Roisin M, Cramer E, Mantha S, et al. A review of outbreaks of foodborne disease associated with passenger ships: evidence for risk management. Public Health Rep 2004; 119:427–34.
- 6. Ortega YR, Sanchez R. Update on cyclospora cayetanensis, a food-borne and waterborne parasite. Clin Microbiol Rev 2010; 23:218–34.
- 7. Centers for Disease Control and Prevention. Guide to Confirming an Etiology in Foodborne Disease Outbreak 2015. Table B-3. Guidelines for Confirmation of Foodborne-Disease Outbreaks (Parasitic). http://www.cdc.gov/foodsafety/outbreaks/investigating-outbreaks/confirming_diagnosis.html (accessed July 3, 2016).
CUTANEOUS, MUCOCUTANEOUS, AND VISCERAL LEISHMANIASIS
Common names for cutaneous disease (CL) are Baghdad or Delhi boil, oriental sore, or espundia in the Old World and uta, chiclero ulcer, or forest yaws in the New World.1 Mucocutaneous (MCL) disease is primarily a disease of South America. Visceral leishmaniasis (VL) is also commonly known as kala-azar. The distribution of leishmaniasis is divided into Old World or the Eastern Hemisphere and New World or the Western Hemisphere (Figures 26.3, 26.4, and 26.5).
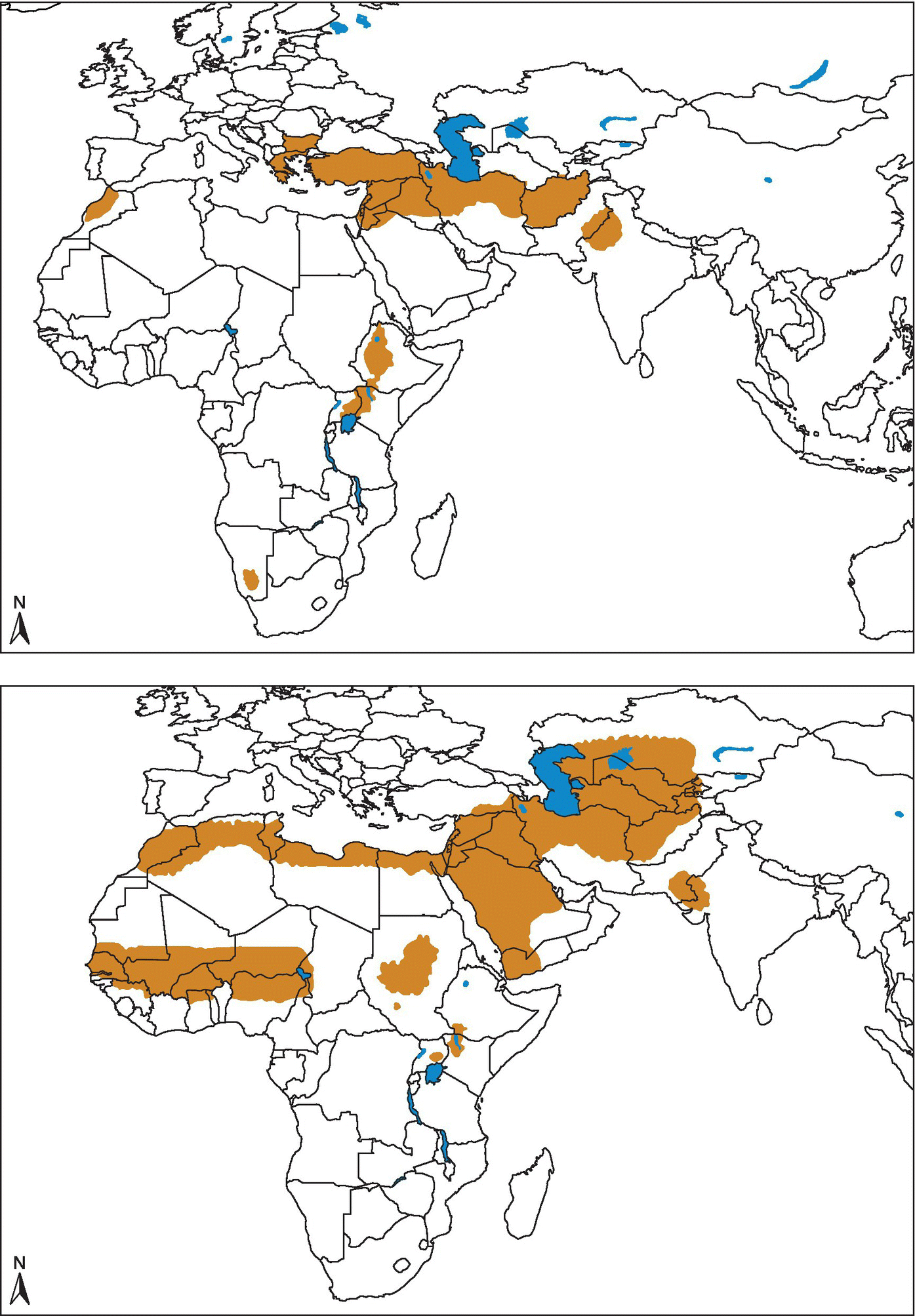
FIGURE 26.3 Cutaneous leishmaniasis. Geographical distribution of Old World cutaneous leishmaniasis due to L. tropica and related species and L. aethiopica. Geographical distribution of Old World cutaneous leishmaniasis due to L. major.
Source: World Health Organization http://www.who.int/leishmaniasis/leishmaniasis_maps/en/ Reprinted with permission.

FIGURE 26.4 Geographical distribution of cutaneous and mucocutaneous leishmaniasis in the New World.
Source: World Health Organization http://www.who.int/leishmaniasis/leishmaniasis_maps/en/ Reprinted with permission.

FIGURE 26.5 Geographical distribution of visceral leishmaniasis in the Old and New world.
Source: World Health Organization http://www.who.int/leishmaniasis/leishmaniasis_maps/en/ Reprinted with permission.
Cutaneous leishmaniasis (CL) is the most common form of leishmaniasis with about 95% of CL cases occurring in the Americas, the Mediterranean basin, and the Middle East and Central Asia. Over two thirds of CL new cases occur in six countries: Afghanistan, Algeria, Brazil, Colombia, the Islamic Republic of Iran, and the Syrian Arab Republic.
Visceral leishmaniasis (VL) occurs in rural tropical and subtropical areas, with over 90% of (VL) cases occurring in Bangladesh, Brazil, Ethiopia, India, South Sudan, and Sudan. VL has been reported in US veterans deployed to Iraq, Kuwait, and Afghanistan.
Almost 90% of MCL cases occur in the Plurinational State of Bolivia, Brazil, and Peru.2
Occupational setting
Leishmaniasis is endemic in 88 countries and found in all continents except Australia and Antarctica. Two million new cases occur yearly—1.5 million cases of CL and MCL and 500 000 cases of VL. The odds of encountering cases of leishmaniasis in nonendemic countries is rising because of an increase in travel by the public sector, occupational exposure through military deployments to disease-endemic areas, and factors that change the pathogenicity of the disease, such as immunodeficiency states (e.g., HIV).3,4 In the Western Hemisphere CL is found in parts of Mexico, Central America, and South America. It is not found in Chile, Uruguay, or Canada. In the United States occasional cases have been reported in Texas and Oklahoma.5 In the Western Hemisphere, workers in forested areas or workers living next to or working next to forested areas—loggers, road builders, agricultural workers, hunters, explorers, scientists, missionaries, and military personnel undergoing jungle training—are at risk. Individuals living and working in farming communities in newly cleared forest areas and workers in road construction and mining are also at risk.1 Workers who spend months in the forests of southern Mexico collecting chewing gum latex, “chicleros,” have a high incidence of infection—30% during the first year of employment.1
For those who are born in geographic areas of Old World CL, the disease is usually a childhood disease, and adults have acquired immunity and are not affected.1 American Regular, Reserve, and National Guard troops and civilian support personnel who have been deployed to Iraq, Kuwait, and Afghanistan have been exposed to leishmaniasis. In 2004 over 600 cases of CL and 4 cases of VL were diagnosed in American soldiers. As these soldiers return to their communities and civilian life, the presentation, diagnosis, and treatment of leishmaniasis have become a challenge for civilian physicians.6
Ecotourists, adventure travelers, bird watchers, and Peace Corps volunteers to endemic areas are also at risk.5
There are other factors that are linked to the risk of occupational exposure.
Poor socioeconomic conditions are a risk factor because poor housing and sanitary conditions (e.g., lack of waste management, open sewerage) may increase sandfly breeding and resting sites. A diet lacking protein energy, iron, vitamin A, and zinc increases the risk that an infection will progress to kala-azar. Epidemics of both CL and VL are often associated with population migration and the movement of nonimmune people into areas with existing transmission cycles. Occupational exposure of people who settle or work in areas that used to be forests and sandfly habitats can lead to an increase in cases. Similarly environmental changes that affect the incidence of leishmaniasis include urbanization, domestication of the transmission cycle, and the incursion of agricultural farms and settlements into forested areas. Leishmaniasis is very climate sensitive and can be affected by changes in rainfall, temperature, and humidity. Small fluctuations in temperature can affect the developmental cycle of Leishmania promastigotes in sandflies, allowing transmission of the parasite in areas not previously endemic for the disease.
Drought, famine, and flood resulting from climate change can lead to massive displacement and migration of people to areas with transmission of leishmaniasis, and poor nutrition could compromise their immunity.2
Exposure (route)
Humans acquire cutaneous leishmaniasis (CL), VL, and MCL through the bite of an infected female phlebotomine sandfly. Sandflies typically feed (bite) at night and during twilight hours and are less active during the hottest time of the day, though they may bite if they are disturbed, such as when individuals brush against tree trunks or other sites where sandflies are resting. The risk of exposure by sandfly bites is often overlooked because sandflies do not make noise, they are small (approximately one third the size of mosquitoes), and their bites might not be noticed.5
Humans, dogs (wild and domesticated), rodents (gerbils), cattle, bats, birds, lizards, edentates (sloths), and marsupials serve as reservoirs. Human reservoir hosts may remain infectious even after clinical symptoms have resolved. In India humans are the only known reservoir. Besides occupational exposure, transmission by blood transfusions, contaminated needles, congenital transmission, and sexual contact have been reported.1
Current American Red Cross recommendations are that any travelers to Iraq wait 12 months before donating blood and that anyone who has been diagnosed with leishmaniasis cannot ever donate blood.7
Pathobiology
CL and MCL are infections that affect the skin and mucous membranes, respectively; they are caused by the vector-borne intracellular protozoan Leishmania. In Asia, Africa, and southern Europe, the agents are Leishmania tropica, Leishmania major, and Leishmania aethiopica. In the Western Hemisphere, the Leishmania braziliensis complex and Leishmania mexicana cause cutaneous and mucocutaneous lesions. The Leishmania donovani complex can cause single cutaneous lesions in both hemispheres; it also can cause visceral disease.
After the sandfly feeds on an infected host, flagellated forms develop and multiply in the sandfly gut. After 8–20 days, infective parasites are present and can be transmitted to another host during a blood meal. After the parasites are injected, they are taken up by macrophages, where they can become amastigotes. The amastigotes multiply, causing macrophage rupture and leading to further spread to other macrophages (Figure 26.6). When an infected sandfly bites and feeds on exposed skin, a small erythematous papule appears after an incubation period of 1–2 weeks to 1–2 months.

FIGURE 26.6 Leishmaniasis Life Cycle: Leishmaniasis is transmitted by the bite of infected female phlebotomine sandflies. The sandflies inject the infective stage (i.e., promastigotes) from their proboscis during blood meals  . Promastigotes that reach the puncture wound are phagocytized by macrophages and other types of mononuclear phagocytic cells. Progmastigotes transform in these cells into the tissue stage of the parasite (i.e., amastigotes) , which multiply by simple division and proceed to infect other mononuclear phagocytic cells . Parasite, host, and other factors affect whether the infection becomes symptomatic and whether cutaneous or visceral leishmaniasis results. Sandflies become infected by ingesting infected cells during blood meals (
. Promastigotes that reach the puncture wound are phagocytized by macrophages and other types of mononuclear phagocytic cells. Progmastigotes transform in these cells into the tissue stage of the parasite (i.e., amastigotes) , which multiply by simple division and proceed to infect other mononuclear phagocytic cells . Parasite, host, and other factors affect whether the infection becomes symptomatic and whether cutaneous or visceral leishmaniasis results. Sandflies become infected by ingesting infected cells during blood meals ( ,
,  ). In sandflies, amastigotes transform into promastigotes, develop in the gut
). In sandflies, amastigotes transform into promastigotes, develop in the gut  (in the hindgut for leishmanial organisms in the Viannia subgenus; in the midgut for organisms in the Leishmania subgenus), and migrate to the proboscis
(in the hindgut for leishmanial organisms in the Viannia subgenus; in the midgut for organisms in the Leishmania subgenus), and migrate to the proboscis  .
.
Life cycle image and information courtesy of DPDx. Source: http://www.cdc.gov/parasites/leishmaniasis/biology.html
All forms of leishmaniasis have a range of clinical disease. In cutaneous leishmaniasis, the scope varies from a progressive nonhealing lesions associated with anergy (diffuse cutaneous leishmaniasis) to the exaggerated hypersensitivity seen in mucosal leishmaniasis and leishmaniasis recidivans, where severe tissue damage is a result of the immune response. In visceral leishmaniasis, many infections are subclinical and self-healing, although malnutrition or immunosuppressive process can reactivate latent infection. In those who develop the syndrome of visceral leishmaniasis, there is suppression of delayed hypersensitivity specifically to leishmanial antigens and nonspecifically to tuberculin and other unrelated antigens. There is also an increase of reticuloendothelial cells and an amplified humoral immune response with the production of nonprotective polyclonal immunoglobulins.1
In cutaneous leishmaniasis, eventually, the papule becomes a nodule, and then an ulcer with characteristic firm, raised, and reddened edges. The ulcer can be dry with a central crust or it may weep seropurulent fluid. It is not usually painful. Subcutaneous nodules may develop along lymphatics, but they represent collections of infected macrophages, not lymph nodes.
Most Old World cutaneous leishmaniasis is caused by infections with L. tropica and L. major. It is characterized by chronic slow-to-heal ulcerative and nodular skin lesions that heal spontaneously, with scarring, after several weeks to several months to as long as 1 year later.1
CL of the New World is caused by parasites of the species complexes L. mexicana and L. braziliensis. The site and appearance of the lesion may be specific for an occupational work group and a geographic region. Chiclero ulcer occurs in Central American forest workers who harvest chicle gum from plants and characteristically develop CL from L. mexicana on the pinna of the ear.1
Mucocutaneous leishmaniasis (MCL), also known as espundia, occurs when parasites from cutaneous lesions metastasize and cause destructive lesions in the oronasopharynx. MCL is found in Brazil, Bolivia, Ecuador, Peru, and other countries of northern and central South America. The most common cause is the L. braziliensis complex. MCL usually occurs after the initial cutaneous lesion has healed, often as long as several years later. The nose is commonly involved, and initial symptoms include stuffiness and intermittent nosebleed. Tissue destruction can involve just the nasal septum, or it can destroy the nose. The upper lip, soft and hard palate, and larynx can also be involved. The parasites in MCL may be difficult to find; in spite of the extensive tissue involvement, VL can have many manifestations. The onset can be acute, subacute, or chronic. The incubation period ranges from weeks to months, but an asymptomatic infection can develop clinical signs and symptoms years to decades after the exposure in people who become immunocompromised for other medical reasons (such as HIV/AIDS). Visceral leishmaniasis usually is caused by the species L. donovani and Leishmania infantum (L. chagasi is considered synonymous with L. infantum) and affects internal organs—particularly the spleen, liver, and bone marrow.
Clinical signs and symptoms of visceral infection include fever, weight loss (cachexia, wasting), hepatosplenomegaly (the spleen is often more prominent than the liver), pancytopenia, and a high total protein level and a low albumin level with hypergammaglobulinemia.
Lymphadenopathy may be seen, particularly in some geographic regions, such as Sudan. HIV-coinfected patients can have atypical manifestations, such as involvement of the gastrointestinal tract and other organ systems. The Hindu term kala-azar—which means black (kala) fever (azar) in Hindi—refers to severe or advanced cases of visceral leishmaniasis. Untreated severe cases of visceral leishmaniasis are often fatal directly from the disease or indirectly from complications, such as secondary mycobacterial infection or hemorrhage.8
The syndrome post-kala-azar dermal leishmaniasis (PKDL) refers to a condition characterized by erythematous or hypopigmented macules, papules, nodules, and patches of the skin. The skin lesions appear first on the face and develop at varying intervals during or after therapy for VL. PKDL is best described in cases of L. donovani infection in South Asia and East Africa. PKDL is more common, develops earlier, and is less chronic in patients in East Africa. In Sudan, PKDL is noted in up to 60% of patients, typically from 0 to 6 months after therapy for visceral leishmaniasis, and often heals spontaneously, while in South Asia, PKDL is noted in ~5–15% of patients several years after initial therapy and usually requires additional treatment. Persons with chronic PKDL can serve as important reservoir hosts of infection.8
Diagnosis
No single diagnostic test for leishmaniasis will give a definitive answer in all clinical settings. The diagnosis is made by finding Leishmania parasites (or DNA) in tissue specimens—such as from scrapings or biopsies of skin lesions for CL or from bone marrow for visceral leishmaniasis. Techniques include light microscopic examination of stained slides, specialized culture techniques, or molecular methods.
Identification of the Leishmania species also can be important, particularly if more than one species is found where the patient lived or traveled since they can have different clinical and prognostic implications. The species can be identified by molecular methods and biochemical techniques (isoenzyme analysis of cultured parasites).
For VL, serologic testing can provide supportive evidence for the diagnosis. The performance of various serologic assays may vary by geographic region and by host factors (e.g., the sensitivity of serologic testing generally is lower in HIV-coinfected patients, particularly if the HIV infection predated the Leishmania infection). Most serologic assays do not reliably distinguish between active and quiescent infection.8
No leishmanin skin-test preparation has been approved for use in the United States.
In the United States, CDC provides reference diagnostic services for leishmaniasis.1,8 Clinicians seeing current or past US military members can find instructions for obtaining help at the following Walter Reed Army Institute of Research (WRAIR) website.9
Treatment
Treatment can produce clinical outcomes (absence of clinical signs or findings) or a parasitologic endpoint cure (the eradication of parasites). The initial clinical response may be followed by relapse requiring retreatment. Treatment choices are often based on the particular parasite strain, the area where the infection occurred, host factors such as a normal or abnormal immune system, and special treatment groups such as young children, elderly persons, and pregnant/lactating women. Specific treatment of leishmaniasis should be individualized and discussed with experienced experts.
Most clinical cases of visceral leishmaniasis and mucosal leishmaniasis should be treated. Even though not all cases of cutaneous leishmaniasis require treatment since many clinically heal on their own over months to years, many CL cases are treated to avoid scarring or the risk of becoming MCL for cases acquired in the New World. Systemic chemotherapy in CL is usually indicated for New World and Old World patients with large or multiple lesions. 1,8
Currently treatment with sodium stibogluconate (Pentostam), a pentavalent antimonial, speeds the time to healing. The entavalent antimonial (SbV) compound sodium stibogluconate (Pentostam®) for IV use is available to US-licensed physicians through the CDC Drug Service (404-639-3670), under an Investigational New Drug (IND) protocol approved by the Food and Drug Administration (FDA) and by CDC’s institutional review board.
Liposomal amphotericin B (AmBisome®), which is administered by IV infusion, is FDA approved for the treatment of visceral leishmaniasis. In 2014, FDA approved the oral agent miltefosine for the treatment of cutaneous, mucosal, and visceral leishmaniasis caused by particular Leishmania species in adults and adolescents at least 12 years of age who weigh at least 30 kg (66 lb). Some medications that might have merit for treating selected cases of leishmaniasis are available in the United States, but the FDA-approved indications do not include leishmaniasis. Examples of such medications include the parenteral agents amphotericin B deoxycholate and pentamidine isethionate, as well as the orally administered “azoles” (ketoconazole, itraconazole, and fluconazole).
Some cases of cutaneous leishmaniasis without risk for mucosal dissemination/disease might be candidates for local therapy, in part depending on the number, location, and characteristics of the skin lesions. Examples of local therapies that might have utility in some settings include cryotherapy (with liquid nitrogen), thermotherapy (use of localized current field radio-frequency heat), intralesional administration of pentavalent antimonial (SbV) compounds (not covered by CDC’s IND protocol for Pentostam®), and topical application of paromomycin (such as an ointment containing 15% paromomycin/12% methylbenzethonium chloride in soft white paraffin, not commercially available in the United States).8
Prevention
Successful prevention and control of leishmaniasis require a combination of strategies because transmission occurs in a complex biological environment involving the human host, parasite, sandfly vector, and in some causes an animal reservoir. Key elements of prevention and control strategies include early diagnosis and effective case management, vector control, effective disease surveillance, control of reservoir hosts, and mobilization and education of the at-risk communities with effective behavioral change interventions and locally tailored communication strategies. Partnership and collaboration with various stakeholders and other vector-borne disease control programs are critical at levels.2
No vaccine or prophylactic medications to prevent leishmaniasis are available at this time. Preventive measures in endemic areas are based on minimizing contact with sandflies. Personal protective methods include avoiding outdoor activities when sandflies are active (dusk to dawn), covering exposed skin, sleeping under fine mesh mosquito netting (18 holes to the linear inch), using insect repellents containing DEET, and saturating mosquito nets and clothing with permethrin. Sleeping in air-conditioned areas is recommended if possible since the fine mesh mosquito netting can be uncomfortable. The use of fans and ventilators is also helpful because sandflies are weak fliers.
Workers and travelers with the potential for exposure should be educated about the transmission and clinical manifestations of leishmaniasis, as well as control methods for the vector phlebotomines (sandflies). Insecticides with residual activity can be used to control sandfly populations. Sandfly breeding sites should be eliminated, as well as the control of principal animal reservoir-infected dogs and rodents.4
For some endemic geographic areas where leishmaniasis is found in people, infected people are not needed to maintain the transmission cycle of the parasite in nature. The animal reservoir hosts (such as rodents or dogs), along with sandflies, maintain the cycle.
In other parts of the world, infected people are needed to maintain the cycle; this type of transmission (human–sandfly–human) is called anthroponotic. In the Indian subcontinent (South Asia), the transmission of L. donovani is anthroponotic. Here early detection and effective treatment of patients can serve as a control measure, while suboptimal treatment can lead to the development and spread of drug resistance. Because the transmission is between and within homes, spraying dwellings with residual-action insecticides and using bed nets treated with long-lasting insecticides may be protective.8
References
- 1. Magill AJ. Leishmaniasis. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Disease, 9th edn. New York: Elsevier, 2013:739–60.
- 2. WHO. http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed December 1, 2014).
- 3. Crum N, Aronson N, Lederman E, et al. History of U.S. military contributions to the study of parasitic diseases. Mil Med 2005; 170(4 suppl):7–29.
- 4. Pesho E, Wortmann G, Neafie R, et al. Cutaneous Leishmaniasis: Battling the Baghdad Boil. Federal Practitioner website, October 2004. http://www.thoracicsurgerynews.com/fileadmin/qhi_archive/ArticlePDF/FP/021100058.pdf (accessed November 3, 2014).
- 5. Centers for Disease Control and Prevention. Infectious Diseases Related to Travel: Leishmaniasis, Cutaneous. http://wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/leishmaniasis-cutaneous (accessed January 4, 2015).
- 6. Weina P, Neafie R, Wortmann G, et al. Old world leishmaniasis: an emerging infection among deployed US military and civilian workers. Clin Infect Dis 2004; 39:1674–80.
- 7. American Red Cross. Donating Blood: Eligibility Requirements. http://www.redcrossblood.org/donating-blood/eligibility-requirements/eligibility-criteria-alphabetical-listing (accessed January 4, 2015).
- 8. CDC. Parasites – Leishmaniasis. http://www.cdc.gov/parasites/leishmaniasis/ (accessed November 1, 2014).
- 9. WRAIR. Leishmaniasis. http://www.pdhealth.mil/downloads/Leishmaniasis_DS_04272004.pdf (accessed June 20, 2016).
NANOPHYETUS
Common name for disease: Human nanophyetiasis
Occupational setting
Fish handlers working with salmon and trout are at risk from exposure.1
Exposure (route)
Eating raw or incompletely cooked, smoked, or salted salmon or steelhead trout causes most cases of human intestinal infection with Nanophyetus salmincola salmincola. Infections have also been reported from ingestion of raw steelhead trout eggs, as well as from handling infected salmonid fish. Infection has been reported in the Pacific Northwest from Nanophyetus salmincola salmincola and in Siberia from Nanophyetus salmincola schihobalowi.2,3
Pathobiology
This zoonotic disease is caused by the trematode N. salmincola salmincola. Disease usually results from ingestion of raw, undercooked, or undersmoked salmonid fishes. Recently, a case was reported in a biological technician due to hand contamination from handling freshly killed, infected juvenile Coho salmon.4
Nanophyetus salmincola salmincola can also infect dogs through the ingestion of infected raw fish. Although it does not cause clinical disease in dogs, it can be the vector of a rickettsial organism, Neorickettsia helminthoeca, which causes a systemic infection called salmon poisoning of dogs. This infection can be fatal in dogs; however, it does not cause disease in humans.2
When N. salmincola eggs from the adult worm are shed in the feces of fish-eating animals such as raccoons, otters, spotted skunks, coyotes, foxes, herons, and diving ducks, miracidia hatch which then penetrate an intermediate snail host. In the snail, the parasite grows; it is shed from the snail as xiphidiocercaria that can penetrate and encyst in 34 species of fish.4 Salmonid fishes seem to be more susceptible.4 The cycle is completed when fish containing the encysted metacercaria are ingested by another animal, allowing the fluke to mature in the intestine. If humans ingest infected fish, they become definitive hosts.2,4
In humans, the clinical findings can range from no symptoms to abdominal pain, bloating, diarrhea, nausea and vomiting, and fatigue. Symptoms seem to be related to worm burdens.3 Eosinophilia can be present and may be significant.2 Eggs appear in the stool 1 week after eating infected fish. The number of eggs in the stool is related to the number of worms causing the infection.2
In the case of the biological technician who was handling infected Coho salmon and removing the posterior one third of the kidney of each fish, infection occurred by accidental hand-to-mouth ingestion of infectious metacercariae.4
Diagnosis
The diagnosis is made in patients with GI symptoms or unexplained eosinophilia by examining the stool for eggs or mature flukes.
Treatment
Bithionol (50 mg/kg orally on alternate days for a total of two doses), niclosamide (2 g orally on alternate days for a total of three doses), and praziquantel (20 mg/kg three times daily for 1 day) are effective treatments for Nanophyetus infection.3,4 In the series of patients treated with praziquantel, stool examinations done 2–12 weeks after treatment were negative for eggs.3
Medical surveillance
Fish handlers who clean and eviscerate infected salmonid fishes should be monitored for symptoms of diarrhea and eosinophilia.4
Prevention
Workers at risk for exposure should wear gloves and practice regular hand washing and good personal hygiene. Fish viscera should be disposed of safely. Thorough cooking, or freezing at −20°C for 24 hours, inactivates metacercarial cysts. Individuals should be advised to avoid eating incompletely cooked, salted or smoked, or raw salmon or steelhead trout.
References
- 1. Dieckhaus KD, Garibaldi RA. Occupational infections. In: Rom WN, ed. Environmental Occupational Medicine, 3rd edn. Philadelphia: Lippincott-Raven, 1998:768.
- 2. Eastburn RL, Fritsche TR, Terhune CA Jr. Human intestinal infection with Nanophyetus salmincola from salmonid fishes. Am J Trop Med Hyg 1987; 36:586–91.
- 3. Fritsche TR, Eastbum RL, Wiggins LH, et al. Praziquantel for treatment of human Nanophyetus salmincola (Troglotrerna salmincola) infection. J Infect Dis 1989; 160:896–9.
- 4. Harrel LW, Deardorff TL. Human nanophyetiasis: transmission by handling naturally infected coho salmon (Oncorhynchus kisutch). J Infect Dis 1990; 161:146–8.
PFIESTERIA PISCICIDA
Common names for disease: Possible estuary-associated syndrome (PEAS)
In the autumn of 1996, watermen (commercial fisherman) reported seeing fish with “punched-out” skin ulcers and erratic swimming behavior in the Pocomoke and neighboring estuaries on the eastern shore of the Chesapeake Bay, Maryland. Sightings continued and increased in the spring and summer of 1997.1,2
Watermen in Maryland who had environmental exposure to water from the affected waterways began reporting learning and memory difficulties. Other complaints included headache, skin lesions, and skin burning on contact with water. A study of 24 exposed individuals showed a dose-related reversible clinical syndrome consisting of difficulties with learning and memory. The symptoms resolved in 3–6 months after stopping exposure.2 The affected waterman had had high-level occupational exposure to waterways where the one-celled dinoflagellated (alga) Pfiesteria piscicida had been identified in association with several fish kill events.
No other reports of clusters of disease attributed to P. piscicida or other Pfiesteria-like organisms (PLOs) have been reported.3 A team of North Carolina medical specialists investigated 67 persons exposed to fish kill waters in North Carolina and found no evidence of adverse health effects from their exposure.3
Since there is no evidence for a diffusible Pfiesteria spp. toxin causing fish death in nature, there is no biomarker of exposure, and the cause and effect relationship of P. piscicida and P. piscicida-related illness in humans remains speculative. Pfiesteria piscicida has been found in waters where there were no reports of symptoms or findings in fish in the waters or in persons exposed to the waters.
In January 1998, a CDC-sponsored work group suggested using the term “possible estuary-associated syndrome” (PEAS) for the possible human health problems that may occur after exposure to estuarine waters. The work group developed specific surveillance criteria (see Diagnosis).3–5
Research after the 1997 event has led to an alternate cause for the fish kill. The dinoflagellate Karlodinium veneficum was also present in Pocomoke in 1997. Karlodinium veneficum has a worldwide distribution and has been implicated in numerous fish kill events around the world since 1950. Late summer fish kills associated with K. veneficum blooms on the Corsica River, an eastern shore subestuary of the Chesapeake Bay, occurred in 2005 and 2006. A K. veneficum toxin has been isolated, its structure has determined, and it has been quantified at specific fish kill events. An alternate explanation for the fish kill events of 1997 and subsequent similar events attributed to Pfiesteria spp. is the co-occurrence of K. veneficum and Pfiesteria spp. The second explanation does not explain the cause(s) of the human health effects related to these events.5
Occupational setting
Fisherman and crabbers (watermen), environmental workers, and laboratory workers who come in contact with P. piscicida toxins in water from river or estuaries during periods of “fish kills” or when fish with Pfiesteria-like lesions are present may be at risk.
Exposure (route)
Skin contact with water in affected waterways and exposure to aerosolized spray from affected waters are the routes of exposure.6
Pathobiology
The relationship of PEAS to the dinoflagellate species and the toxins has not been fully characterized. It is not clear that the “clinical neurotoxic” symptoms are directly due to the toxins produced by P. piscicida or Pfiesteria-like dinoflagellates.2 It is unknown whether individuals exposed to P. piscicida while swimming, boating, or engaging in other types of recreational activities in coastal waters are at risk for developing PEAS. PEAS does not appear to be infectious, since there is no association with the consumption of fish or shellfish caught in waters containing P. piscicida.4 The evidence that suggests a relationship between PEAS and toxins produced by P. piscicida is based on the reports that individuals exposed to estuarine water in Maryland prior to and during fish kills associated with P. piscicida toxin developed symptomatic neurocognitive deficits. All deficits resolved by 3–6 months after stopping exposure.2,3
While there is a report of learning difficulties in laboratory rats associated with exposure to water from aquaria containing P. piscicida toxins (2,3), a study designed and conducted under the guidance of an independent expert Task Force on Health Risk of Exposure to Fish Kill Waters by a team of medical specialists from North Carolina found no evidence of severe, chronic, or widespread adverse health effects from exposures to fish kill waters in North Carolina.3
The lack of a definitive link between Pfiesteria species and their toxins with criteria for PEAS in humans has been the result of both the difficulties in the field identification of these organisms and the species complexity of local algal blooms. 5 While Pfiesteria spp. can now be detected and identified in water and sediment samples and also at fish kill events, there has been no indication of involvement of Pfiesteria spp. in any fish kill event since 1998.
Diagnosis
The presence of P. piscicida does not indicate risk to fish or humans. Fish lesions can also result from a variety of biological physical and environmental factors. CDC, along with other federal, state, and local government agencies and academic institutions, has set up organized surveillance for PEAS.4 The surveillance system tracks PEAS rather than P. piscicida-related illness, since P. piscicida toxin has not been linked to the criteria for PEAS.5,7
The CDC PEAS criteria are:
- Developing symptoms within 2 weeks after exposure to estuarine water.
- Reporting memory loss or confusion of any duration and/or three or more selected symptoms (i.e., headache, skin rash at the site of water contact, sensation of burning skin, eye irritation, upper respiratory irritation, muscle cramps, and GI symptoms) that—with the exception of skin rash at the site of water contact and sensation of burning skin—persist for 2 or more weeks.
- A healthcare provider cannot identify another cause for the symptoms.4
Treatment
Due to the lack of evidence of the specific cause of symptoms, current treatment is avoiding further exposure. During the initial description of PEAS, there are case reports of treatment with cholestyramine, a toxin-binding polymer.8
Medical surveillance
Since 1998 there has been no indication of the involvement of P. piscicida in any fish kill event. Individuals who experience health problems or providers who see patients with symptoms after exposure to estuarine water, a fish disease event, or a fish kill site should contact their healthcare provider and their state or local public health agency.
Prevention
Even though no definitive cause or toxin has been identified, from a commonsense point of view, individuals who are occupationally exposed to fish kill waters should avoid areas with diseased, dying, or dead fish or shellfish. Persons should not go in or near water in areas that have been closed by governmental agencies and should not harvest or eat fish or shellfish from such areas.3,4 People who come in contact with water from fish kill areas should wash well with plain soap and water and wash any clothes that come in contact with such water. Use waterproof gloves when handling items or objects contaminated with fish kill waters.
References
- 1. Centers for Disease Control and Prevention. Results of the public health response to Pfiesteria workshop—Atlanta, Georgia, September 29–30, 1997. MMWR 1997; 46:951–2.
- 2. Grattan LM, Oldach D, Tracy JK, et al. Learning and memory difficulties after environmental exposure to waterways containing toxic-producing Pfiesteria or Pfiesteria-like dinoflagellates. Lancet 1998; 352:532–9.
- 3. Smith CG, Music SI. Pfiesteria in North Carolina: the medical inquiry continues. N C Med J 1998; 59:216–9.
- 4. CDC. Possible estuary-associated syndrome. MMWR 1999; 48:381–2.
- 5. Robledo JAF, Deeds JR, Place JR, et al. Unresolved questions and an alternative hypothesis. In: Botana LM, ed. Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection, 2nd edn. Boca Raton: CRC Press/Taylor & Francis Group, 2008:717–51.
- 6. Warrell DA. Fish poisoning: gastrointestinal and neurotoxic syndromes. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases, 9th edn. New York: Elsevier, 2013:925–7.
- 7. CDC. Surveillance for possible estuary-associated syndrome (Pfiesteria), 6 states. MMWR 2000; 49:372–3.
- 8. Shoemaker RC, Hudnell HK. Possible estuary-associated syndrome: symptoms, vision and treatment. Environ Health Perspect 2001; 109(5):539–45.
PLASMODIUM SPECIES
Common name for disease: Malaria
Occupational setting
Malaria is a serious worldwide disease. 3.4 billion people live in areas at risk of malaria transmission in 106 countries and territories. The majority (56%) of malaria infections are in sub-Saharan Africa, followed by Southeast Asia (27%), the Eastern Mediterranean (12%), and South America (3%).1 The World Health Organization estimates that in 2012 malaria caused 207 million clinical episodes and 627 000 deaths. An estimated 91% of deaths in 2010 were in the African region.2
Workers or travelers in endemic areas are at risk from exposure to malaria through bites from mosquitoes. Occupational factors that increase the risk of malaria include working in rural areas where mosquito control methods are not effective or nonexistent, working during mosquito biting hours (dusk to dawn for Anopheles sp.), and working without protective clothing or insect repellents.
One interesting, although uncommon, occupational exposure is airport malaria. Airport malaria refers to malaria case reports of individuals who never traveled to malarious areas and who also lacked other risk factors for malaria, such as a history of blood transfusions or intravenous drug abuse. In several reported cases, the victims worked near or at an international airport and were thought to have been infected by the bite of an infected tropical Anopheles sp. mosquito when cabin or cargo hold doors were opened. During the summer of 1994, six cases of airport malaria were found around Roissy Charles de Gaulle Airport.3 Four of the cases were in airport workers and two other cases lived in Villeparisis about 7.5 km away. The nonairport worker cases were thought to be from Anopheles mosquitoes that were carried in the cars of airport workers who lived next door. Hot, humid summer weather is thought to be a factor that allows the survival of infected Anopheles mosquitoes brought by airplanes.2
A 2002 review found 89 cases of airport malaria between 1969 and 1989.4 Airport malaria does not include cases in persons who became infected during brief stops at airports in malaria-endemic areas, nor does it include those who may have acquired the disease from an infected Anopheles sp. mosquito during a flight.5
Human population movement is a significant factor in the reemergence of malaria worldwide. Besides the intercontinental movement of infected people from endemic areas to nonmalarious areas through modern transportation, population movement may lead to environmental changes that increase the risk of malaria by creating better habitats for Anopheles mosquitoes. Examples are deforestation, the creation of irrigation systems, and rapid, unregulated urbanization accompanied by poor housing and sanitation.2
Malaria was eradicated from the United States in the 1940s. However, 1400–1900 cases are reported to CDC each year—with 1925 cases in 2011.6 The largest number of cases is typically seen in New York City and other ports of entry (Figure 26.7). The majority of cases are acquired during international travel. This group includes business travelers, pleasure travelers, active duty military members serving in endemic areas, and first- and second-generation immigrants (including their spouses) who travel back to their country of origin to visit friends and relatives (VFR travelers). While 75% of the cases are associated with failure to use recommended prophylaxis, approximately half of all cases of malaria in US travelers are among VFR travelers.2

FIGURE 26.7 Number of malaria cases by state or territory in which case was diagnosed—United States, 2011. Number of malaria cases (N = 1925) by state or territory in which case was diagnosed—United States, 2011. Nearly all of these cases are imported, though there are rare instances of transmission due to blood transfusion or organ transplantation, congenital transmission, or mosquito-borne cases. Approximately 3–5 malaria deaths occur annually in the U.S. Most of these deaths are due to delayed diagnosis and treatment.
Source: http://www.cdc.gov/mmwr/preview/mmwrhtml/ss6205a1.htm.
VFR travelers as a group have many factors that place them at greater risk for getting malaria. Their duration of travel tends to be longer than for other types of travel such as business trips or tourist travel tours. They also are more likely to stay at the houses of these friends and relatives versus hotels, and based on the destination or socioeconomic status, the private homes may be less likely to be air conditioned or have screened windows. VFR travelers are less likely to use the recommended malaria prevention measures such as insect repellent and chemoprophylactic medicines. Reasons for this behavior include socioeconomic factors such as access to healthcare or health insurance. The lack of access can limit appropriate preventive medical interventions including malaria prophylaxis. VFR travelers often consider themselves to be at low or no risk for infection because they grew up in a malaria-endemic country and consider themselves to be immune. Part of this mistaken perception is the belief that even if they are infected, the infection will be mild and can be easily treatable with medicines they can acquire while abroad.
Any partial immunity that VFR travelers may have developed while growing up in a malaria-endemic country is lost very quickly after moving away making them as vulnerable to infection as people who grew up in nonendemic countries. Likewise their children and spouses who may be accompanying them on the trip will also have no immunity. The medicines that are available overseas to treat malaria may not be appropriate or effective and may not meet the same quality standards as those found in the United States. VFR travelers need to follow the same preventive measures that are recommended for all travelers.2
Rarely, cases in the United States occur through exposure to infected blood products, by congenital transmission or by local mosquito-borne transmission.6
Exposure (route)
Infected female Anopheles sp. mosquitoes transmit malaria. The malaria sporozoite is introduced into humans when the mosquito punctures the skin to feed. Malaria transmission is definitely influenced by climate. Sporogony does not occur at temperatures below 16°C or higher than 33°C. The optimal conditions are between 20 and 30°C, and the mean relative humidity is at least 60%.7
Pathobiology
Historically, four species of Plasmodium parasites were considered capable of infecting humans:
Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae. Recently, a fifth species, Plasmodium knowlesi, that causes malaria among monkeys and occurs in certain forested areas of Southeast Asia has been recognized as a significant human pathogen.7
The life cycle of the Plasmodium sp. that cause human malaria involves humans and the Anopheles sp. mosquito. The mosquito becomes infected after feeding on an infected human. If Plasmodium gametocytes of both sexes are present, the sexual cycle leads to the creation of an ookinete (a mobile, fertilized egg). The ookinete penetrates through the gut, where an oocyst forms. After 2 weeks, this oocyst ruptures, releasing sporozoites. Many of these make their way into the mosquito’s salivary glands. An infected female Anopheles sp. mosquito is then capable of inoculating malaria sporozoite forms into humans while she feeds. The sporozoites rapidly enter the bloodstream and within hours enter liver cells (hepatocytes) and develop into liver-stage schizonts. After an asexual multiplication stage, each shizont ruptures, releasing 10 000–40 000 uninucleate merozoites, which can invade red blood cells.
Inside the red blood cell, each merozoite develops asexually before rupturing the red blood cell, releasing more merozoites and continuing the cycle. The red blood cell cycle takes 48–72 hours, depending on the species. Plasmodium falciparum, P. vivax, and P. ovale take 48 hours, and P. malariae takes 72 hours. The one exception is P. knowlesi whose cycle is 24 hours. The red blood cell stages of the malaria parasite are primarily asexual. Occasional merozoites become male or female gametocytes, permitting the life cycle to continue.6 Both P. vivax and P. ovale can have dormant forms of the parasites, hypnozoites, which can remain in the liver and become liver schizonts months or years after the initial inoculation. The liver schizonts can start new cycles (relapses) of red blood cell infections—an important fact when treating malaria caused by these two species 7(Figure 26.8).

FIGURE 26.8 Malaria parasite life cycle. The malaria parasite life cycle involves two hosts. During a blood meal, a malaria-infected female Anopheles mosquito inoculates sporozoites into the human host . Sporozoites infect liver cells  and mature into schizonts
and mature into schizonts  , which rupture and release merozoites
, which rupture and release merozoites  . (Of note, in P. vivax and P. ovale a dormant stage [hypnozoites] can persist in the liver and cause relapses by invading the bloodstream weeks, or even years later.) After this initial replication in the liver (exo-erythrocytic schizogony
. (Of note, in P. vivax and P. ovale a dormant stage [hypnozoites] can persist in the liver and cause relapses by invading the bloodstream weeks, or even years later.) After this initial replication in the liver (exo-erythrocytic schizogony  ), the parasites undergo asexual multiplication in the erythrocytes (erythrocytic schizogony
), the parasites undergo asexual multiplication in the erythrocytes (erythrocytic schizogony  ). Merozoites infect red blood cells
). Merozoites infect red blood cells  . The ring stage trophozoites mature into schizonts, which rupture releasing merozoites
. The ring stage trophozoites mature into schizonts, which rupture releasing merozoites  . Some parasites differentiate into sexual erythrocytic stages (gametocytes)
. Some parasites differentiate into sexual erythrocytic stages (gametocytes)  . Blood stage parasites are responsible for the clinical manifestations of the disease. The gametocytes, male (microgametocytes) and female (macrogametocytes), are ingested by an Anopheles mosquito during a blood meal . The parasites’ multiplication in the mosquito is known as the sporogonic cycle
. Blood stage parasites are responsible for the clinical manifestations of the disease. The gametocytes, male (microgametocytes) and female (macrogametocytes), are ingested by an Anopheles mosquito during a blood meal . The parasites’ multiplication in the mosquito is known as the sporogonic cycle  . While in the mosquito's stomach, the microgametes penetrate the macrogametes generating zygotes
. While in the mosquito's stomach, the microgametes penetrate the macrogametes generating zygotes  . The zygotes in turn become motile and elongated (ookinetes)
. The zygotes in turn become motile and elongated (ookinetes)  which invade the midgut wall of the mosquito where they develop into oocysts
which invade the midgut wall of the mosquito where they develop into oocysts  . The oocysts grow, rupture, and release sporozoites
. The oocysts grow, rupture, and release sporozoites  , which make their way to the mosquito’s salivary glands. Inoculation of the sporozoites into a new human host perpetuates the malaria life cycle.
, which make their way to the mosquito’s salivary glands. Inoculation of the sporozoites into a new human host perpetuates the malaria life cycle.
Plasmodium falciparum is the species most commonly associated with severe disease. Plasmodium vivax is the dominant species found outside of Africa (the Middle East, Asia, the Western Pacific, and Central and South America). Plasmodium ovale is found in West Africa. While P. malariae has a worldwide distribution, it is usually only found in isolated pockets. Plasmodium knowlesi so far is restricted to South and Southeast Asia, usually in areas harboring macaque monkeys. Statistically P. falciparum and P. vivax are the most common, and P. falciparum causes the most deaths.1,7
Even though most symptoms occur within 2 weeks of exposure and 95% of cases occur within 6 weeks of inoculation, an occasional case of P. vivax may not develop symptoms for years after exposure.7 In a person with no prior exposure to malaria, the presenting symptoms go through four successive stages of chills, fever, sweats, and remission of fever. Nonspecific symptoms associated with malaria include headache, back pain, muscle pain, and malaise. When partial immunity has developed due to repeated exposure, the symptoms of infection may be much milder or, on occasion, nonexistent.7
Malaria is classically described with cyclical episodes of fever every 3 days (tertian) for P. falciparum, P. vivax, and P. ovale and every 4 days (quartan) for P. malariae. Fever follows the release of blood merozoites from rupture of erythrocytes after schizogony. The cyclical fevers are only evident when a population of parasites predominates and the majority of the schizogony ends cyclically and in synchronization.
Plasmodium falciparum causes the most severe complications. It can cause neurologic symptoms, from headaches to seizures and cerebral edema. Plasmodium falciparum can also cause pulmonary edema, glomerulonephritis, renal failure, liver dysfunction, anemia, and hypoglycemia. Plasmodium vivax, P. ovale, and P. malariae can lead to anemia, splenomegaly, and headache; however, they generally do not cause the severe symptoms and mortality associated with P. falciparum.7
Patients diagnosed with malaria are generally categorized as having either uncomplicated or severe malaria. Patients diagnosed with uncomplicated malaria can be effectively treated with oral antimalarials. The diagnosis of severe malaria is made when the patient has one or more of the following clinical criteria: impaired consciousness/coma, severe normocytic anemia [hemoglobin < 7], renal failure, acute respiratory distress syndrome, hypotension, disseminated intravascular coagulation, spontaneous bleeding, acidosis, hemoglobinuria, jaundice, repeated generalized convulsions, and/or parasitemia of ≥5%. Patients with severe disease should be treated aggressively with parenteral antimalarial therapy.1
While individuals of all ages remain susceptible to infection, immunity to severe disease can develop over time, from repeated exposure to bites from infected anopheline mosquitoes. While antidisease immunity is not fully understood, transmission intensity seems to be a major factor. Young children in sub-Saharan Africa, tourists, and soldiers in other malaria-endemic areas are considered to be nonimmune and are at risk of developing severe and complicated malaria. Semi-immune people can be infected (i.e., parasitemic) but can be asymptomatic. If semi-immune people develop a malaria illness, their only symptoms may be fever and malaise. They rarely develop symptoms of severe life-threatening malaria.7
Pregnant women have an increased susceptibility to P. falciparum malaria. Pregnant women who have partial immunity due to repeated infections may lose some of their immunity because of immune system changes during pregnancy and the presence of the placenta and additional place where malaria parasites can bind.3
In malaria-endemic countries, P. falciparum contributes to 8–14% of low birth weight, which decreases the chance of a baby’s survival.3
Diagnosis
Patients suspected of having malaria infection should be urgently evaluated. Suspicion should lead to a search for parasites in Giemsa-stained blood smears. Thick blood smears contain 20–40 times more blood than thin smears and are more sensitive. Thin blood smears are better for species identification once malaria parasite forms have been seen. A negative blood smear makes the diagnosis of malaria unlikely. However, because nonimmune individuals may be symptomatic at very low parasite densities that may be undetectable by blood smear, blood smears should be repeated every 12–24 hours for a total of three sets. If all three are negative, the diagnosis of malaria has been essentially ruled out.2
After malaria parasites are detected on a blood smear, the parasite density should be estimated. The thin smear can be used to identify the species, since it is important to determine if the infection is due to P. falciparum, since this species causes the greatest morbidity and mortality and is often resistant to chloroquine. Identifying P. vivax and P. ovale is also important, because they require treatment of the hypnozoites in the liver to prevent relapses, as well as treatment of the blood-stage infection.2 Mixed cases of malaria can also occur.
Besides microscopy, other laboratory diagnostic tests that are available are rapid diagnostic tests (RDT), polymerase chain reaction (PCR) tests, and serologic tests.8 RDTs can more rapidly determine that the patient is infected with malaria, but they cannot confirm the species or the parasitemia. PCR is more sensitive and specific than microscopy, but it can only be performed in reference laboratories—a disadvantage since it may take time to get the results from a reference lab. An advantage of PCR is that it is a very useful tool for confirmation of species and detecting of drug resistance mutations. Serologic tests, which are also only performed in reference laboratories, can assess past malaria experience but not current infection. CDC offers malaria drug testing for all malaria diagnosed in the United States at no charge.2
Treatment
While malaria should be considered in anyone who has recently traveled in a malaria-endemic area who presents with a fever, whether they took prophylaxis or not, the treatment for malaria should not be started until the diagnosis of malaria has been definitely established. Treatment should be based on three main factors: the infecting Plasmodium species, the clinical status of the patient, and the drug susceptibility of the infecting parasites based on the geographic area where the infection was acquired and the previous use of antimalarial medicines.2
Identifying the Plasmodium species for treatment purposes is important because P. falciparum and P. knowlesi infections can rapidly lead to progressive severe illness or death, while the other species, P. vivax, P. ovale, or P. malariae, are less likely to cause severe manifestations. Plasmodium vivax and P. ovale infections also require treatment for the hypnozoite forms that remain dormant in the liver and can cause a relapsing infection. Finally, P. falciparum and P. vivax species have different drug resistance patterns in differing geographic regions. For P. falciparum and P. knowlesi infections, the urgent initiation of appropriate therapy is especially critical.
The clinical status of the patient—whether they have uncomplicated or severe malaria—determines whether they can be treated with oral antimalarials or in the case of severe malaria should be treated aggressively with parenteral antimalarial therapy.
Knowledge of the geographic area where the infection was likely acquired provides information on the chances of drug resistance of the infecting parasite. Considering this information the treating clinician can choose an appropriate drug or drug combination. If a malaria infection occurred despite use of a specific medicine for chemoprophylaxis, that medicine should not be a part of the treatment regimen. If faced with the clinical situation where the diagnosis of malaria is suspected and cannot be confirmed or if the diagnosis of malaria is confirmed but species determination is not possible, antimalarial treatment effective against chloroquine-resistant P. falciparum should be started immediately.2
The treatment choice should rapidly reduce or eliminate the parasitemia, and in the case of severe malaria it should prevent the complications of severe malaria. Drug therapy must reduce or eliminate the asexual blood stage and in patients with P. vivax or P. ovale must also include a drug such as primaquine, which eliminates the liver stage, to prevent relapses.6 Treatment decisions should include input from an infectious disease or tropical disease expert or discussion with CDC for specific treatment regimens. (CDC clinicians are on call 24 hours to provide advice to clinicians on the diagnosis and treatment of malaria and can be reached through the Malaria Hotline 770-488-7788 (or toll free 855-856-4713) Monday–Friday, 9:00 a.m. to 5:00 p.m. On off-hours, weekends, and federal holidays, call 770-488-7100 and ask to have the malaria clinician on call to be paged.)
The current medications for the treatment of malaria are active against the parasite forms in the blood and include:
- Chloroquine
- Atovaquone/proguanil (Malarone®)
- Artemether/lumefantrine (Coartem®)
- Mefloquine (Lariam®)
- Quinine
- Quinidine
- Doxycycline (used in combination with quinine)
- Clindamycin (used in combination with quinine)
- Artesunate (not licensed for use in the United States, but available through the CDC malaria hotline)
Patients who have severe P. falciparum malaria or who cannot take oral medications should be given the treatment by intravenous infusion. The current intravenous drugs regimens are quinidine gluconate plus one of the following: doxycycline, tetracycline, clindamycin, or artesunate (see above), followed by one of the following: atovaquone/proguanil (Malarone®), doxycycline (clindamycin in pregnant women), or mefloquine. Consultation with an infectious disease or tropical disease expert or discussion with CDC for specific treatment regimens is recommended.2 While primaquine is active against the dormant parasite liver forms (hypnozoites) and prevents relapses, it should not be taken by pregnant women or by people who are deficient in glucose-6-phosphate dehydrogenase (G6PD). Primaquine can cause severe hemolysis in persons with low activity of G6PD, and patients should undergo G6PD testing before receiving the medication. Primaquine should not be given to pregnant women, because the drug may cross the placenta to a G6PD-deficient fetus and lead to hemolysis in utero.2,7
Prevention
Prevention strategies include avoidance and control of the mosquito vector, as well as chemoprophylaxis for travel to endemic areas. Chemoprophylaxis should always be used in conjunction with personal protective measures to avoid mosquito bites. Individuals can minimize their chance of contact with the Anopheles sp. mosquito by using barrier methods that include wearing long-sleeve shirts and full-length pants, sleeping under insecticide-treated bed nets, staying in accommodations with screened windows or air conditioning, and avoiding exposure by staying indoors, especially at dusk and dawn when mosquitoes are most active (Figure 26.9).

FIGURE 26.9 How travelers can protect themselves against malaria. This picture shows some things that travelers can use to protect themselves against malaria: malaria pills, insect repellent, long-sleeved clothing, bed net, and flying insect spray. (Not shown, but also protective: air conditioned or screened quarters.) Based on the risk assessment, specific malaria prevention interventions should be used by the traveler. Often this includes avoiding mosquito bites through the use of repellents or insecticide treated bed nets, and specific medicines to prevent malaria.
Personal protective measures include the use of mosquito repellents containing the following ingredients: DEET up to 50% (products containing DEET include Off!, Cutter, Sawyer, and Ultrathon); picaridin, also known as KBR 3023, Bayrepel, and icaridin (products containing picaridin include Cutter Advanced, Skin So Soft Bug Guard Plus, and Autan [outside the United States]); oil of lemon eucalyptus (OLE) or PMD (products containing OLE include Repel and Off! Botanicals); and IR3535 (products containing IR3535 include Skin So Soft Bug Guard Plus Expedition and SkinSmart). Spraying rooms with pyrethrum-containing spray and spraying clothing with permethrin are other methods to minimize mosquito contact.2
Vector control methods include reducing the breeding areas of mosquitoes by draining or eliminating standing water and by killing the adult and (larval) stages of the mosquito.
Individuals traveling to malaria-endemic areas should see a healthcare provider with expertise in travel medicine for a travel evaluation and risk assessment. The risk assessment, including the need for antimalarial prophylaxis, should take into account the destination country; detailed itinerary, including specific cities, types of accommodation, season, the style of travel, and length of stay; age; pregnancy and breastfeeding status; current medications; and general medical history. Additional considerations when choosing an antimalarial include the following: Does the traveler have a medical condition (including, but not limited to, G6PD deficiency for chloroquine and primaquine, renal impairment for atovaquone/proguanil, or seizure disorders for mefloquine) or allergy that would be a contraindication for certain antimalarials? Does the traveler currently take any medication that might interact with certain antimalarials? Does the traveler have preferences that may affect adherence (a daily vs. a weekly medication)? Is cost of medication a consideration for the patient? Are there logistical issues? Prophylactic drugs need to be started prior to leaving for the trip—anywhere from 1 day to 2 weeks prior depending on the medication.2 Commonly used drugs for chemoprophylaxis include atovaquone/proguanil (Malarone®), chloroquine, doxycycline, mefloquine (Lariam®), and primaquine.2 CDC maintains country-specific tables that include resistance patterns and current recommendations.2 These should be consulted prior to providing a prescription.
A final aspect of malaria prevention is the necessity for prompt diagnosis and treatment of malarial disease. Ninety percent of all travelers who develop malaria do not show symptoms until they return home. The number of cases of P. falciparum infections from travel to sub-Saharan Africa continues to increase, even though it represents less than 2% of all travel. The increase in cases is due to the high risk of malaria to travelers in both urban and rural areas of sub-Saharan Africa. Malaria can still occur in persons who use personal protective measures and take chemoprophylaxis correctly.
In the United States malaria transmitted through blood transfusion is very rare with a rate of less than 1 per 1 million units of blood transfused. Prevention of transfusion cases of malaria depends on collecting a travel history from presenting donors and following FDA screening guidelines:
- Most travelers to an area with malaria are deferred from donating blood for 1 year after their return.
- Former residents of areas where malaria is present will be deferred for 3 years.
- People diagnosed with malaria cannot donate blood for 3 years after treatment, during which time they must have remained free of symptoms of malaria.2
References
- 1. Taylor T, Agbenyega T. Malaria. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Disease, 9th edn. New York: Elsevier, 2013:695–717.
- 2. Centers for Disease Control and Prevention. Travelers Health: Malaria. http://wwwnc.cdc.gov/travel/diseases/malaria (accessed January 5, 2015).
- 3. Martens P, Hall L. Malaria on the move: human population movement and malaria transmission. Emerg Infect Dis 2000; 6:103–9.
- 4. Guillet P, Germain MC, Chandre F, et al. Origin and prevention of airport malaria in France. Trop Med Int Health 1998; 3:700–5.
- 5. Thang HD, Elsas RM, Veenstra J. Airport malaria: report of a case and a brief review of the literature. Neth J Med 2002; 60(11):441–3.
- 6. Isaacson M. Airport malaria: a review. Bull WHO 1989; 67:737–43.
- 7. Centers for Disease Control and Prevention. Malaria surveillance, 2011. MMWR 2013; 62(SS-5):1–18.
- 8. World Health Organization. Malaria: Diagnostic Testing. http://www.who.int/malaria/areas/diagnosis/en/ (accessed January 5, 2015).
TOXOPLASMA GONDII
Common name for disease: Toxoplasmosis
Occupational setting
Toxoplasmosis is caused by a single-celled parasite called Toxoplasma gondii. It was considered to be benign in immunocompetent persons and potentially severe in immunocompromised patients and for the fetus. More recent research shows that the clinical spectrum and severity of disease depend on the host and the pathogen’s genetic background, the immune status of the host, and the host–parasite interaction. Toxoplasmosis is now recognized as a possible etiology for a severe infectious syndrome in travelers living in, or returning from, tropical countries.1,2
The parasite is found throughout the world, but the incidence depends on local eating habits, environmental conditions, and the presence of definitive hosts. Prevalence increases with age. In Western Europe the prevalence is 30–50%. In France, the prevalence declined from more than 80% in the 1960s to 54.3% in 1995 and to 43.8% in 2003.1 The past high prevalence of infection in France has been related to a preference for eating raw or undercooked meat, particularly lamb; and the high prevalence in Central America has been related to the frequency of stray cats in a climate favoring the survival of oocysts and soil exposure. Reasons for the declines are believed to be improved hygiene, more widespread freezing of meat—which kills cysts—and better awareness in the general population regarding the risks of consuming undercooked meat. In the United States the seroprevalence has declined from 14.1% in the 1988–1994 National Health and Nutrition Examination Survey (NHANES) to 9% in the 1999–2004 NHANES.1 Even with the decline in the United States, toxoplasmosis is estimated to be the second leading cause of death from foodborne illness and the fourth leading cause of foodborne illness hospitalization.3,4
When a person with a healthy immune system becomes infected, their immune system usually keeps the parasite from causing illness. In pregnant women and individuals who have compromised immune systems, Toxoplasma infection can cause serious health problems.
Occupations such as butchers, slaughterhouse workers, laboratory workers (occupational exposure and transmission by needlestick can occur when working with live Toxoplasma), landscapers, gardeners, farmers, veterinarians and their associates, pet store owners, and cat breeders are at risk from exposure.1,3,5 In the United States a case–control study of adults recently infected with T. gondii, working with meat, was associated with recent T. gondii infection. The authors postulated that some persons who work with meat could inadvertently ingest undercooked meat (e.g., if gloves were not worn or were removed incorrectly). They also suggested that cooks who work with meat could taste dishes before the meat was fully cooked or could inadvertently ingest undercooked meat from their hands.6
Workers in stables or grain elevators where feral or house cats are present also can be exposed.7 Seronegative missionaries, volunteers, and workers for agencies in endemic areas are also at risk for toxoplasmosis.
Exposure (route)
Human infection is usually through contact with and ingestion of parasite oocysts from:
- Eating undercooked, contaminated meat especially pork, lamb, and venison
- Accidental ingestion of undercooked, contaminated meat after handling it and not washing hands thoroughly (Toxoplasma cannot be absorbed through intact skin.)
- Eating food that was contaminated by knives, utensils, cutting boards, and other foods that have had contact with raw, contaminated meat
- Drinking water contaminated with T. gondii or drinking unpasteurized milk (goat) contaminated with T. gondii
- Accidentally ingesting the parasite through contact with cat feces that contain Toxoplasma. This can happen by:
- Cleaning a cat’s litter box when the cat has shed Toxoplasma in its feces
- Touching or ingesting anything that has come into contact with cat feces that contain Toxoplasma
- Accidentally ingesting contaminated soil (e.g., not washing hands after gardening or eating unwashed fruits or vegetables from a garden)
Other mechanisms are mother-to-child (congenital) transmission and rarely by receiving an infected organ transplant or infected blood by transfusion.3
The only known definitive hosts for T. gondii are members of family Felidae (domestic cats and their relatives). A cat that has eaten a single infected bird or mouse can greatly increase the risk to other animals and humans by shedding millions of oocysts in its feces during the 2-week period of the intestinal phase of its infection. The oocysts can survive for more than a year in moist soil. Most people who become infected do not have symptoms, and those who do become may only have mild illness often with only swollen lymph nodes.3,5
However, recently illness with more severe symptoms has been seen in immunocompetent travelers to South America (French Guiana) and Africa.2,3,8
Pathobiology
Members of the cat family (Felidae) serve as the definitive host and do not require an intermediate host to become infected. Cats become infected after consuming intermediate hosts harboring tissue cysts or become directly infected by ingestion of sporulated oocysts. One unique characteristic of the infection is the transmission of infection from one intermediate host to another by carnivorism, without need for passage through a definitive cat host. Domestic animals bred for human consumption and wild game may also become infected with tissue cysts after ingestion of sporulated oocysts in the environment. Common intermediate hosts are pigs, sheep, deer, rabbits, rats, mice birds, cattle, and humans.1
Toxoplasma gondii can exist in three forms: the tachyzoite and the trophozoite forms of acute infections; the bradyzoite, found in the tissue cyst; and the oocyst, shed in cat feces. Oocysts transform into tachyzoites shortly after ingestion and multiply rapidly during the acute infection. Tachyzoites localize in the neural and muscle tissue and are the cause of acute illness. They destroy their host cells, enter adjacent cells, and result in focal necrosis and a vigorous local inflammatory reaction. Tachyzoites divide every 5–12 hours and can cause significant cell destruction until immunity is acquired. After 3–6 weeks, the infection persists with only slowly dividing bradyzoites inside tissue cysts. Neither chemotherapy nor host immunity affects the parasite once this transformation occurs. The long-lived bradyzoites persist inside cysts during the remainder of the hosts’ life and are responsible for latency and reactivation. Toxoplasma infection is the presence of the trophozoite form or the cyst form in tissues whether there is clinical disease or not. The tachyzoite is present in the host tissues during the acute stage of the infection. Tissue cysts containing the bradyzoites develop in host tissues and are the main mode of transmission of the organism to carnivores, including humans.1 The oocyst is produced only through an enteroepithelial cycle in the intestines of the cat family. Cats are important in the spread of toxoplasmosis, because they can deposit massive numbers of oocysts that remain infective for months. Cats develop a primary infection during their first year of life, when they begin hunting and ingest tissue cysts in rodents, birds, or oocysts from other cat feces. Kittens and cats can shed millions of oocysts in their feces for as long as 3 weeks after infection, and the oocysts can remain infective for over a year. Mature cats are less likely to shed Toxoplasma if they have been previously infected.3 The oocysts sporulate 1–5 days after excretion. Oocysts are easily spread by wind and rain or on fomites. Oocysts tend to survive longer in moist, shaded soil. Their survival is decreased under temperature extremes, low humidity, and exposure to direct sunlight. The common chemical disinfectants are not effective in inactivating oocysts. Once sporulated oocysts or tissue cysts are ingested, the extraintestinal cycle can occur. Trophozoite forms are released from the oocysts. They invade the GI mucosa and reach all organs by means of the bloodstream. Within the organs, the organisms invade and kill cells (Figure 26.10).

FIGURE 26.10 Toxoplasma gondii Life Cycle: The only known definitive hosts for Toxoplasma gondii are members of family Felidae (domestic cats and their relatives). Unsporulated oocysts are shed in the cat’s feces . Although oocysts are usually only shed for 1–2 weeks, large numbers may be shed. Oocysts take 1–5 days to sporulate in the environment and become infective. Intermediate hosts in nature (including birds and rodents) become infected after ingesting soil, water or plant material contaminated with oocysts . Oocysts transform into tachyzoites shortly after ingestion. These tachyzoites localize in neural and muscle tissue and develop into tissue cyst bradyzoites . Cats become infected after consuming intermediate hosts harboring tissue cysts . Cats may also become infected directly by ingestion of sporulated oocysts. Animals bred for human consumption and wild game may also become infected with tissue cysts after ingestion of sporulated oocysts in the environment . Humans can become infected by any of several routes:
- eating undercooked meat of animals harboring tissue cysts .
- consuming food or water contaminated with cat feces or by contaminated environmental samples (such as fecal-contaminated soil or changing the litter box of a pet cat) .
- blood transfusion or organ transplantation .
- transplacentally from mother to fetus .
In the human host, the parasites form tissue cysts, most commonly in skeletal muscle, myocardium, brain, and eyes; these cysts may remain throughout the life of the host. Diagnosis is usually achieved by serology, although tissue cysts may be observed in stained biopsy specimens . Diagnosis of congenital infections can be achieved by detecting T. gondii DNA in amniotic fluid using molecular methods such as PCR .
Source: http://www.cdc.gov/parasites/toxoplasmosis/biology.html
Differentiation of tachyzoites into bradyzoites correlates with the onset of protective immunity. When the immune system gains control of the infection, the organism encysts and remains as a tissue cyst in the host. In the setting of T-cell immune deficiency, bradyzoites can reactivate into tachyzoites. The genetic background of the host may influence the risk of reactivation. In HIV-infected patients the risk of reactivated toxoplasmosis is higher or lower depending on the host’s human leukocyte antigen (HLA) haplotype.1 About 80% of immunocompetent persons who become infected by sporulated oocysts are asymptomatic. Among the 20% who develop symptoms, the incubation period is 1–2 weeks, and symptoms can resemble a flu-like illness.
When symptoms develop, the most common are fever and transient head and neck lymphadenopathy, most commonly of the posterior cervical nodes. Other systemic nonspecific symptoms can include fatigue, sore throat, myalgia, and headache. The usual time course for symptoms is 2–4 weeks. The disease is usually self-limited and resolves spontaneously within a few weeks or months. The differential diagnosis includes infectious mononucleosis and lymphoma. Occasionally, patients develop myocarditis, splenomegaly, encephalitis, hepatomegaly, and retinochoroiditis. When antibody appears, it results in lifelong immunity against reinfection. Tissue cysts are not eradicated by antibody, and maintenance of an antibody titer can be an indicator of the continuing presence of live organisms in the cysts.1 Cases of severe primary toxoplasmosis caused by an atypical or recombinant strain of T. gondii in immunocompetent patients have been reported. The majority of patients were infected in French Guiana, but cases have also been reported from other areas. Some of the European cases are linked to undercooked horsemeat imported from South America. The clinical presentation is prolonged fever higher than 39°C, weight loss of >5% body weight, generalized lymphadenopathy, elevated liver enzyme levels, and lung involvement. Fifty percent of the patients have severe headache, and a third have diarrhea, hepatosplenomegaly, or chorioretinitis. Without treatment, this severe presentation is often fatal.1
Differentiation of tachyzoites into bradyzoites correlates with the onset of protective immunity and begins after a few days. In the setting of T-cell immune deficiency, bradyzoites can reactivate into tachyzoites. The genetic background of the host may have influence the risk of reactivation. This is well known in HIV-infected patients for whom the risk of reactivated toxoplasmosis is higher or lower depending on the host’s human leukocyte antigen (HLA) haplotype. The absence of effective T-cell immunity in the fetus explains the gravity of congenital toxoplasmosis.
In immunocompromised patients, toxoplasmosis is a severe disease, and most cases are reactivation of chronic infection resulting from impaired of T-cell immunity. High-risk groups include HIV-infected patients with a CD4 cell count <100/mm, patients treated with immunosuppressive agents, and bone marrow transplant patients.
The most common presentation is encephalitis and another common clinical presentation is pneumonia (which can resemble pneumocystosis), but any organ can be involved because T. gondii infects all nucleated cell types. Disseminated toxoplasmosis usually presents with fever, altered mental status, arthralgia, rash, and focal findings, such as CNS abscess, pneumonia, or elevated liver enzymes.1
Severe generalized toxoplasmosis with high fatality rates occurs when recipient seronegative persons receive donor positive organs. Universal prophylaxis with trimethoprim/sulfamethoxazole (TMP/SMX) is effective in preventing posttransplant toxoplasmosis.1
Ocular toxoplasmosis is the leading cause of posterior uveitis worldwide. In the past it was believed to be a late consequence of congenital toxoplasmosis; the current thought is that the majority of ocular disease in immunocompetent patients is from postnatally acquired infection. Ocular involvement can be linked with recent primary infection and is more frequently reported in South America, particularly Brazil.1 It can also be delayed several months or even years after primary infection.1
PREGNANT WOMEN
A woman who is newly infected with Toxoplasma during pregnancy can pass the infection to her unborn child (congenital infection). The woman may not have symptoms, but there can be severe findings in the unborn child, such as diseases of the nervous system and eyes.3 The rate of transplacental infection and the severity of disease in the fetus are related to the time of infection during gestation. First trimester maternal infection carries a risk of fetal infection that is only about 10%, but the resulting disease is severe. Third trimester maternal infection increases the risk of fetal infection to 65% and approaches 100% at term, but the neonatal infection is usually asymptomatic.1
Diagnosis
The diagnosis of acute toxoplasmosis can be made by the serologic pattern, a finding of trophozoites in tissue, isolation of trophozoites from blood or body fluids, lymph node histology, or a finding of cysts in tissue.1
The Sabin–Feldman dye test is very specific and sensitive and is considered the gold standard for detecting Toxoplasma antibodies in humans, but it creates a potential occupational hazard because it requires the use of live parasites. Since so many cases are asymptomatic, the dye test is not helpful in distinguishing recent from past infection, since the titer peaks early and remains elevated. It is currently done only in select reference laboratories.9
IgG and IgM antibody (Ab) tests by IFA or by ELISA are available. The IgG Ab test is specific and sensitive, with levels peaking in 1–2 months and persisting for several months or years. The IgM antibodies are the first to be detected and are useful in diagnosing acute infection (1,3). IgG Ab levels are helpful in determining the prevalence of infection, the risk of reactivation in the immunocompromised, and the risk of acquiring Toxoplasma infection. Diagnosis of acute acquired infection can be based on seroconversion, a fourfold rise, two-tube rise in IgG antibody titer, or the presence of a high level of IgM antibody. Because they do not show a rise in titer, diagnosis of reactivated infection in immunocompromised patients is more difficult.1
Molecular techniques that can detect the parasite’s DNA in the amniotic fluid can be useful in cases of possible mother-to-child (congenital) transmission.
Ocular disease is diagnosed based on the appearance of the lesions in the eye, symptoms, course of disease, and often serologic testing.3
Treatment
Most of the medications used to treat toxoplasmosis are active only against the tachyzoite forms of the parasite and do not eradicate the infection. Atovaquone does have limited activity against the cyst forms.1
The most common side effect is dose-related suppression of the bone marrow, which may be decreased by concomitant administration of folinic acid (calcium leucovorin).10
Lymphadenopathic toxoplasmosis symptoms in immunocompetent adults are usually self-limited. With visceral disease that is clinically evident or if symptoms are severe or persistent, treatment may be indicated for 2–4 weeks. Pyrimethamine is considered to be the most effective agent which should be included in drug regimens used against the parasite. Pyrimethamine is a folic acid antagonist. Leucovorin protects the bone marrow from the toxic effects of pyrimethamine. A second drug, such as sulfadiazine or clindamycin, should be added.10 Other medications include a fixed combination of trimethoprim with sulfamethoxazole as well as less extensively studied drugs such as atovaquone and pyrimethamine plus azithromycin. Corticosteroids are sometimes prescribed along with antiparasitic agents.3
For ocular diseases treatment should be based on a complete ophthalmologic evaluation, and then the decision to treat ocular disease is dependent on factors such as acuteness of the lesion, degree of inflammation, visual acuity, lesion size, location, and persistence.3
Management of maternal and fetal infection varies and should be discussed with infectious disease specialists. Spiramycin is often recommended for the first and early second trimesters or pyrimethamine/sulfadiazine and leucovorin for late second and third trimesters for women with acute T. gondii infection diagnosed at a reference laboratory during gestation. PCR is often performed on the amniotic fluid at 18 gestation weeks to determine if the infant is infected. If the infant is likely to be infected, then treatment with pyrimethamine, sulfadiazine, and leucovorin is typical. Congenitally infected newborns are usually treated with pyrimethamine, sulfonamide, and leucovorin.
Persons with AIDS who develop active toxoplasmosis (usually toxoplasmic encephalitis) need treatment along with antiretroviral therapy to significantly improve their immunologic status.3
Medical surveillance
Surveillance in areas or countries with a high prevalence of Toxoplasma may be appropriate for workers in specific high-risk settings listed above and particularly for women of childbearing age. Initial screening may include IgG and IgM antibodies. Periodic screening can be done annually. Seronegative women at high risk of acquiring toxoplasmosis who become pregnant should be screened early in pregnancy, again at 20–22 weeks, and near or at term to find those who may have acquired primary infections during pregnancy.1 Another alternative would be to offer removal from potential exposure to all seronegative pregnant women for the duration of their pregnancy.
In China T. gondii serological surveys done from the late 1990s to the early to mid-2000s in various provinces show a range of 25–45% positivity in slaughterhouse workers, 10–24% positivity in dairy workers, 12.5–26% positivity in veterinarians, 14–21% positivity in meat-processing workers, 10–30% positivity in cooks, and 12–20% positivity in animal breeders.11
Prevention
Currently there is no vaccine against toxoplasmosis and there is no justification for chemoprophylaxis. Prevention for people at risk is education about the organism, the routes and symptoms of infection, and the oocysts in the environment. For those working outdoors, gloves should be worn in environments where fecal contamination may be found. Workers should avoid touching facial mucous membranes with gloves.
Prevention is most important in seronegative pregnant women and immunodeficient patients. Again it is most readily accomplished through education.
Specific educational points include:
- Avoid contact with materials potentially contaminated with cat feces, especially handling of cat litter and gardening. Gloves are advised when these activities are necessary. Because oocysts require 1–2 days to mature, dispose of all cat feces daily.
- Keep outdoor sandboxes covered.
- Feed cats only canned or dried commercial food or well-cooked table food, not raw or undercooked meats.
- Change the litter box daily if you own a cat. The Toxoplasma parasite does not become infectious until 1–5 days after it is shed in a cat’s feces.
- Since epidemiology studies have identified water as a potential source for T. gondii infection both in humans and animals, river and lake water should be boiled prior to drinking.
- Cook food to safe temperatures, and since tachyzoites cannot survive desiccation, freezing and thawing freeze meat for several days at subzero (0°F) temperatures before cooking greatly reduce chance of infection.
- Meat that is smoked, cured in brine, or dried may still be infectious.
- Peel or wash fruits and vegetables thoroughly before eating.
- Wash countertops, cutting boards, dishes, counters, utensils, and hands with hot soapy water after contact with raw meat, poultry, seafood, or unwashed fruits or vegetables.
- Avoid drinking untreated drinking water.
- Wear gloves when gardening and during any contact with soil or sand because it might be contaminated with cat feces that contain Toxoplasma. Wash hands with soap and warm water after gardening or contact with soil or sand.
- Teach children the importance of washing hands to prevent infection.1,10
If you are pregnant or immunocompromised:
- Avoid changing cat litter if possible. If no one else can perform the task, wear disposable gloves and wash your hands with soap and warm water afterward.
- Keep cats indoors.
- Do not adopt or handle stray cats, especially kittens. Do not get a new cat while you are pregnant.3
On farms, seronegative pregnant women should avoid in lambing activities. Cats should not be used to control rodents; traps or rodenticides should be used instead. Cats should not be allowed inside feed-storage facilities, and all cat feces should be removed from buildings and stalls. Farm cats should be fed palatable dry or canned food, not forced to forage and hunt. Pigs or cats should never be permitted to eat uncooked scraps of pork or sheep. Dead animals should be removed promptly to prevent cannibalism.1
References
- 1. Paris L. Toxoplasmosis. In: Magill AJ, Hill DR, Solomon T, et al., eds. Hunter’s Tropical Medicine and Emerging Infectious Disease, 9th edn. New York: Elsevier, 2013:765–75.
- 2. Anand R, Jones CW, Ricks JH, et al. Acute primary toxoplasmosis in travelers returning from endemic countries. J Travel Med 2012; 19:57–60.
- 3. Centers for Disease Control and Prevention. Parasites: Toxoplasmosis. http://www.cdc.gov/parasites/toxoplasmosis/ (accessed January 5, 2015).
- 4. Jones LJ, Praise ME, Fiore AE. Neglected parasitic infections in the United States: toxoplasmosis. Am J Trop Med Hyg 2014; 90:794–9.
- 5. The Canadian Centre for Occupational Health and Safety (CCOHS). Toxoplasmosis. http://www.ccohs.ca/oshanswers/diseases/toxoplasmosis.html (accessed January 5, 2015).
- 6. Teutsch SM, Juranek DD, Sulzer A, et al. Epidemic toxoplasmosis associated with infected cats. N Engl J Med 1979; 300:695–9.
- 7. Jones JL, Dargelas V, Roberts J, et al. Risk factors for Toxoplasma gondii infection in the United States. Clin Infect Dis 2009; 49(6):878–84.
- 8. Khan A, Ajzenberg D, Mercier A, et al. Geographic separation of domestic and wild strains of Toxoplasma gondii in French Guiana correlates with a monomorphic version of chromosome1a. PLoS Negl Trop Dis 2014; 8(9):e3182. doi:10.1371/journal.pntd.0003182. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4169257/pdf/pntd.0003182.pdf (accessed December 14, 2014).
- 9. Udonsom R, Buddhirongawatr R, Sukthana Y. Is Sabin-Feldman dye test using T. gondii tachyzoites from animal inoculation still the best method for detecting Toxoplasma gondii antibodies? Southeast Asian J Trop Med Public Health 2010; 41(5):1059–64.
- 10. Montoya JG, Boothroyd J, Civics JA. Toxoplasma gondii. In: Bennett JE, Dolin R, Blaser MJ, et al., eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th edn., Philadelphia: Saunders/Elsevier, 2015:3122–53.
- 11. Zhou P, Chen Z, Li H, et al. Toxoplasma gondii infection in humans in China. Parasit Vectors 2011; 4:165. doi:10.1186/1756-3305-4-165. http://www.parasitesandvectors.com/content/4/1/165 (accessed December 14, 2014).