
CHAPTER 6
STRUCTURAL SEARCH IN RNA MOTIF DATABASES
6.1 INTRODUCTION
Ribonucleic acid (RNA) is transcribed from deoxyribonucleic acid (DNA) and plays a key role in the synthesis of proteins [1]. An RNA structural motif is a substructure of an RNA molecule that has a significant biological function. Well-known RNA structural motifs include the iron response element (IRE) and histone 3′-UTR stem-loop (HSL3) [2,3]. As increasingly more RNA structural motifs are discovered, it becomes crucial to have databases holding the motifs that can be accessed and used by researchers. For example, Rfam [4] and RNA STRAND [5] are two such databases.
Rfam is a well-annotated, open access database containing information on noncoding RNA (ncRNA) families as well as other RNA structural motifs. The latest version of Rfam 9.0, comprising 603 families in total, is available at http://rfam.sanger.ac.uk/. In Rfam, each ncRNA family is represented by two structure-annotated multiple sequence alignments (MSAs). One MSA is called the seed alignment and the other is called the full alignment. Each multiple sequence alignment is associated with a consensus secondary structure, represented in Stockholm format [6,7]. The seed alignment consists of functionally related RNA sequences obtained from the literature or wet lab experiments.
The seed alignment is used to build a covariance model used by the Infernal program [6] to collect additional functionally related RNA sequences. These additional RNA sequences obtained from the Infernal program are added to the full alignment.
Rfam is equipped with several different search methods. By entering a query sequence, the user can search the covariance models representing the 603 noncoding RNA families in Rfam. Since the computational cost involved with covariance models is high, Rfam employs BLAST as a filter to speed up searches. When a search is completed, the search result shows the RNA families that have a high degree of similarity to the input query sequence. The user can view and browse the RNA families displayed in the search result. In addition to the search-by-sequence method, Rfam allows the user to search the ncRNA families stored in its database via keyword and EMBL ID or accession number. The entire Rfam database can also be downloaded in plain text format from the Rfam website and searched offline locally using the Infernal program [6] on the user’s own computer.
While Rfam provides keyword- and sequence-based search methods, it lacks structure-based search methods. Many functionally related ncRNAs differ at the sequence level but are similar in their secondary structures. Thus, it is desirable to have structure-based search engines on RNA motif databases. This chapter presents two structural search engines developed in our lab. The first search engine is installed on a database, called RmotifDB [8], which contains secondary structures of the ncRNA sequences in Rfam. The second search engine is installed on a block database, which contains the 603 seed alignments, also called blocks, in Rfam. This search engine employs a novel tool, called BlockMatch, for comparing multiple sequence alignments. We report some experimental results to demonstrate the effectiveness of the BlockMatch tool.
6.2 THE SEARCH ENGINE ON RMOTIFDB
This section presents the structure-based search engine on RmotifDB. This search engine employs our previously developed program, called RSmatch [9], for comparing two RNA sequences taking into account their secondary structures. In what follows, we first review the RSmatch tool and then describe RmotifDB and its structural search engine.
6.2.1 RSmatch
Functional RNA motifs may be detected by aligning the secondary structures of RNA sequences on which the motifs exist. Many software tools have been developed to find the RNA motifs by aligning the RNA secondary structures. However, existing software tools suffer from some drawbacks. They either require a large number of prealigned RNA sequences or have high time complexities. Therefore, these tools have difficulty in processing RNAs without prealigned sequences or in handling large RNA structure databases. RSmatch is a software tool designed for comparing RNA secondary structures and for motif detection. The RSmatch algorithm is fast; its time complexity is O(mn), where m is the length of a query RNA structure and n is the length of a subject RNA structure. The algorithm decomposes an RNA secondary structure into a collection of structure components. To capture the structural particularities of RNA, RSmatch uses a tree model to organize these structure components. The tool compares a pair of RNA secondary structures using two separate scoring matrices in performing local and global alignments. One scoring matrix is used for single-stranded regions and the other is used for double-stranded regions. When searching an RNA structure database, RSmatch can detect similar RNA substructures, and perform an iterative database search and multiple structure alignment. These operations enable the identification and discovery of functional RNA structural motifs.
By conducting experiments with instances of known RNA structural motifs, including simple stem-loops and complex structures with junctions, we have demonstrated that in detecting the RNA structural motifs, the accuracy of RSmatch is high compared with other software tools [9]. RSmatch is especially useful to scientists and researchers interested in aligning RNA structural motifs obtained from RNA folding programs or wet lab experiments where the size of the RNA structure data set is large. The software is available from http://datalab.njit.edu/biodata/rna/RSmatch/software.htm. Figure 6.1 presents a screenshot illustrating the execution of RSmatch’s database search function in the Unix command line environment.
FIGURE 6.1 A screenshot showing the execution of RSmatch in Unix.

6.2.2 The RmotifDB System
RSmatch offers an efficient algorithm for aligning two RNA structures, along with a basic RNA database search capability, but it must be run offline on a user’s local machine, which is a major drawback. Even RADAR (http://datalab.njit.edu/biodata/rna/RSmatch/server.htm) [10,11], a descendant of RSmatch with an excellent web interface for aligning two RNA structures, does not contain a search method for a large database. In Section 6.2.1, we observed that there are provisions for sequence and keyword searching in the Rfam database, but not structure searching. To intensively study RNA structural functions or motifs, a structure-based search engine for RNA motif databases is needed. With this motivation, we have built RmotifDB, which is equipped with a structural search engine and is available at http://datalab.njit.edu/bioinfo/singleseq_index.html.
To build the database for RmotifDB, the plain text seed alignment file with 603 ncRNA families is downloaded from the Rfam 9.0 website. A total of 18,233 ncRNA sequences are extracted from this seed alignment file. Each of these sequences is then folded using the Vienna RNA package’s RNAfold [12] to obtain its secondary structure. Finally, the entire 18,233 ncRNA sequences along with their secondary structures are stored in a single plain text file, which constitutes the major database file for RmotifDB. The RSmatch version 2.0 is used by the structural search engine of RmotifDB. The RSmatch 2.0 software is downloaded from the RADAR website. The implementation uses a perl-cgi approach to integrate the system’s web interface with RSmatch. This allows the use of the structural search engine over the web via a browser.
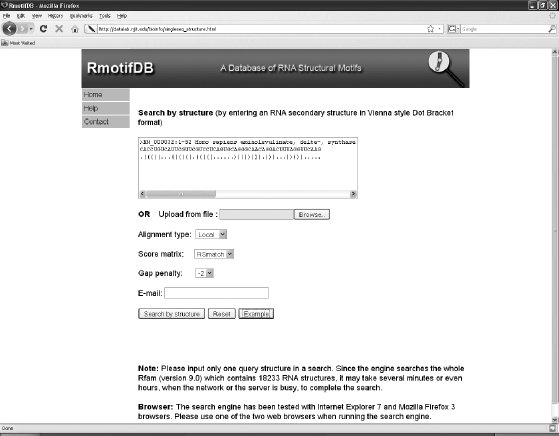
RmotifDB supports searching for “nearest neighbors” of RNA structural motifs from its database. Nearest neighbors of an RNA structural motif are other motifs with a high degree of similarity to the given motif. The two major search modes provided by RmotifDB are search-by-sequence and search-by-structure. The user can submit a query, which is either an RNA sequence or an RNA secondary structure, through RmotifDBs web interface. If the RNA sequence is given, it must be in FASTA format. This sequence is then folded by RNAfold and used to compare with the secondary structures in the database file of RmotifDB. If the RNA structure is given, it must be in the Vienna style Dot Bracket format. The user can either paste the input query into a text box or upload the query from a plain text file. Additional options include variations on the alignment type, the scoring matrix, and the gap penalty. Local or global alignment can be selected as the alignment type. Currently, the only scoring matrix used is RSmatch’s default matrix, but additional options for scoring matrices can be accommodated. The default gap penalty is -2, which can be changed based on the user’s preference. The user’s e-mail address is required. Figure 6.2 illustrates the web interface for the search-by-structure method in RmotifDB.
FIGURE 6.2 The web interface of RmotifDBs search-by-structure method.
On completion of a search, an e-mail notification is sent to the user with a link to the result, in which the subject RNA structures are ranked based on their similarity to the query with the most similar structure being ranked highest. Figure 6.3 shows a search result in RmotifDB. Since the structural search engine uses RSmatch to compare the given query sequence or structure, one by one, with the 18,233 RNA secondary structures in Rfam (version 9.0), it may take minutes or even hours to complete the search when the server is busy.
RmotifDB capitalizes on RSmatch and the Rfam database to build a structure-based search engine. By providing a convenient browser-style interface, RmotifDB
FIGURE 6.3 An example search result in RmotifDB.

provides the ability to search for ncRNA structural motifs via search-by-sequence and search-by-structure methods, benefiting researchers interested in ncRNAs functions and structural motifs.
6.3 THE SEARCH ENGINE BASED ON BLOCKMATCH
BlockMatch is a software tool capable of aligning and comparing two RNA blocks. An RNA block is a structure-annotated multiple RNA sequence alignment represented in Stockholm format. Examples of blocks can be found in Rfam [4], where a seed alignment is a block, or in GLEAN-UTR DB [13], where a group of functionally related RNA sequences is a block. The BlockMatch web server is available at http://datalab.njit.edu/compbio/blockmatch/.
This section presents a block database and the structural search engine on the database. The block database contains the 603 seed alignments, also called blocks, in Rfam. The search engine accepts a user-input query block in Stockholm format, then uses BlockMatch to compare the query block one by one with the seed alignment of each ncRNA family in Rfam, and finally displays the seed alignments that are most similar to the query block. Each seed alignment is a block in Stockholm format. The query block could represent a putative RNA structural motif, and the database search function allows the detection of Rfam families that are closely related to the structural motif. These Rfam families can be considered as structural neighbors of the motif.
6.3.1 Search by Block
The input interface of the structural search engine on the block database is shown in Figure 6.4. The example query block, in Stockholm format, in the figure is called group 3 taken from GLEAN-UTR DB, which is the Histone 3′-UTR stem–loop group found in human messenger RNA (mRNAs). The query block can be pasted into the text box or uploaded from a plain text file. If a file is provided, then the block in the file is used as the query. If no file is uploaded, then the block in the text box is used as the query. The user can change the number of hits he/she wants to display. The default number is 5. When the search result is available, the user will be notified via e-mail.
FIGURE 6.4 The web interface of the structural search engine based on BlockMatch.

Figure 6.5 shows a sample result obtained from the structural search engine on the block database. The structural search engine compares the user-input query block, one by one, with the seed alignments in Rfam using the BlockMatch algorithm, and finds related Rfam families whose seed alignments are most similar to the query block with the largest alignment scores. The search result consists of a summary of the query block including its length and size, (i.e., the number of sequences in the multiple alignment of the query block, the date and time of query submission, and the date and time of the completed search, shown in the top table in Figure 6.5), followed by the Rfam families that are closely related to the query block, with the top ranked family being the most similar to the query block (shown in the bottom table in Fig. 6.5).
FIGURE 6.5 A sample result returned by the structural search engine based on BlockMatch.

6.3.2 Experiments and Results
To our knowledge, the structural search engine based on BlockMatch is the first one of its kind designed for comparing structure-annotated multiple RNA sequence alignments. We have compared this search engine with the RNA structure alignment tool RADAR [10] described in Section 6.3.1. Given a query RNA secondary structure and a database of subject RNA secondary structures, RADAR is able to find the subject structures similar to the query structure.
We conducted a series of experiments to evaluate the relative performance of BlockMatch and RADAR. We first downloaded two blocks from the GLEAN-UTR database [13] accessible from http://datalab.njit.edu/biodata/GLEAN-UTR-DB/. This database contains putative structural motifs in human and mouse UTRs. The first block we downloaded is called group 3 in the database, which is the Histone 3′-UTR stem-loop (HSL3) group in human homolog sequences. The second block we downloaded is called group 9 in the database, which is the Iron response element (IRE) group in human homolog sequences. Group 3 contains 6 HSL3 sequences, denoted by HSL3i, 1 ≤ i ≤ 6. Group 9 contains 6 IRE sequences, denoted by IREi, 1 ≤ i ≤ 6. Table 6.1 lists the two blocks and details of the sequences in each block. Each block contains a multiple alignment of six sequences together with the consensus secondary structure of the sequences.
TABLE 6.1 The HSL3 and IRE Blocks Used in Our Experimental Study.

In the first experiment, we used the Vienna RNA package’s RNAfold [12,14] to fold each sequence in the two blocks in Table 6.1 to obtain 12 secondary structures, and to fold the sequences in Rfam release 9.0 into secondary structures. There were 18,233 RNA sequences grouped into 603 blocks (seed alignments) in the Rfam database, among which there were 64 HSL3 sequences and 39 IRE sequences; hence we obtained 18,233 secondary structures in total for the Rfam database. Each sequence together with its secondary structure in Table 6.1 was used as a query to search the Rfam database by comparing the query with the secondary structures in Rfam, one by one, using RADAR. In the second experiment, we compared each query sequence (structure) in Table 6.1 with the seed alignments or blocks of the 603 families in Rfam using BlockMatch. (In an extreme case, BlockMatch can take a single RNA secondary structure and compare it with a block.) In the third experiment, we compared each block in Table 6.1 with the 18,233 secondary structures in Rfam using BlockMatch. Finally, in the fourth experiment, we compared each block in Table 6.1 with the 603 seed alignments (blocks) in the Rfam database.
The performance measures used to evaluate the performance of the tools are sensitivity, Sn, and specificity, Sp, where
where TP is the number of true positives, FN is the number of false negatives, TN is the number of true negatives, and FP is the number of false positives. There were 64 HSL3 structures and 39 IRE structures in Rfam. In experiments 1 and 3, a true positive is an HSL3 (IRE, respectively) structure that is returned as one of the top 64 (39, respectively) hits by the tools. A false positive is a non-HSL3 (non-IRE, respectively) structure that is returned as one of the top 64 (39, respectively) hits. A true negative is a non-HSL3 (non-IRE, respectively) structure that is not returned as one of the top 64 (39, respectively) hits. A false negative is an HSL3 (IRE, respectively) structure that is not returned as one of the top 64 (39, respectively) hits.
There was a single HSL3 block (accession RF00032) and a single IRE block (accession RF00037) among the 603 blocks in Rfam. In experiments 2 and 4, TP equals 1 if the HSL3 (IRE, respectively) block is ranked as the top hit by BlockMatch, or 0 otherwise. The FP equals 1 if the HSL3 (IRE, respectively) block is not ranked as the top hit, or 0 otherwise. The TN equals 602 if the HSL3 (IRE, respectively) block is ranked as the top hit, or 601 otherwise. The FN equals 1 if the HSL3 (IRE, respectively) block is not ranked as the top hit, or 0 otherwise.
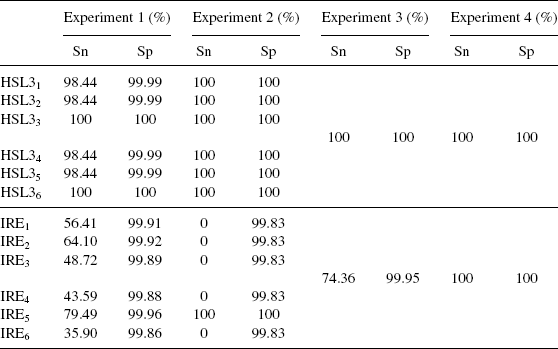
Table 6.2 summarizes the experimental results. Combining all sequences into a query block and using BlockMatch to align the query block with all the blocks in Rfam yielded the best performance, where BlockMatch achieved a sensitivity value of 100% and a specificity value of 100%. In this case, the tool was able to take into account the characteristics of all sequences in the blocks while aligning them. In experiments 2 and 3, where a sequence (structure) was aligned with a block, partial sequence information was lost, and hence the performance of BlockMatch degraded. In experiment 1, where a query sequence (structure) was aligned with each individual sequence (structure) in the Rfam database, the notion of blocks was completely missing and the RADAR tool yielded the worst performance. These experimental results show that when blocks are available, one should use BlockMatch to do block alignments in performing database searches, hence considering all sequences in an entire block during the alignments, as opposed to using single sequence or structure matching tools to align individual sequences or structures in the blocks.
TABLE 6.2 Sensitivity and Specificity Values Obtained from Our Experiments.
Note that BlockMatch computes the alignment score of two blocks by considering both the sequence alignment and the consensus structure in each block. We conducted experiments to evaluate the impact of changing the sequence alignment or consensus structure on alignment results. When the sequence alignment or the consensus structure in a block changes, so does the alignment score. The alignment score depends on how many base pairs and single bases occur and what nucleotides are present in each column of the alignment. Suppose there are four sets of blocks {A1, B1}, {A2, B2}, {A3, B3}, and {A4, B4}. Here A1 and B1 have similar sequences and similar consensus structures; A2 and B2 have similar sequences but different consensus structures; A3 and B3 have different sequences but similar consensus structures; and A4 and B4 have different sequences and different consensus structures. The alignment score between A1 and B1 is generally higher than that of A2 and B2 (A3 and B3, respectively), which is generally higher than the alignment score between A4 and B4.
6.4 CONCLUSION
This chapter presented two structure-based search engines for RNA motif databases. These search engines are useful to scientists and researchers who are interested in RNA secondary structure motifs. The search engine on RmotifDB employs the efficient RSmatch program to perform pairwise alignment of RNA secondary structures. The search engine on the block database uses BlockMatch to perform pairwise alignment of blocks or seed alignments. Both the RNA secondary structures and seed alignments are taken from the widely accessed Rfam database. In future work, we plan to extend the techniques presented here to search for three-dimensional motifs in RNA structure databases [15,16].
ACKNOWLEDGMENTS
We thank Mugdha Khaladkar, James McHugh, Vandanaben Patel, and Bruce Shapiro for helpful conversations while preparing this manuscript.
REFERENCES
1. J. T. L. Wang and X. Wu (2006), Kernel design for RNA classification using support vector machines, Inter. J. Data Mining Bioinformat., 1:57–76.
2. W. F. Marzluff and R. J. Duronio (2002), Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences, Curr. Opin. Cell Biol., 14:692–699.
3. G. Pesole, S. Liuni, G. Grillo, F. Licciulli, F. Mignone, C. Gissi, and C. Saccone (2002), UTRdb and UTRsite: specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs, Nucleic Acids Res., 30:335–340.
4. S. G. Jones, S. Moxon, M. Marshall, A. Khanna, S. R. Eddy, and A. Bateman (2005), Rfam: annotating non-coding RNAs in complete genomes, Nucleic Acids Res., 33:D121–D124.
5. M. Andronescu, V. Bereg, H. H. Hoos, and A. Condon (2008), RNA STRAND: The RNA secondary structure and statistical analysis database, BMC Bioinformat., 9:340.
6. S. R. Eddy (2002), A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure, BMC Bioinformat., 3:18.
7. J. Spirollari, J. T. L. Wang, K. Zhang, V. Bellofatto, Y. Park, and B. A. Shapiro (2009), Predicting consensus structures for RNA alignments via pseudo-energy minimization, Bioinformat. Biol. Insights, 3:51–69.
8. D. Wen and J. T. L. Wang (2009), Design of an RNA structural motif database, Inter. J. Comput. Intell. Bioinformat. Systems Biol., 1:32–41.
9. J. Liu, J. T. L. Wang, J. Hu, and B. Tian (2005), A method for aligning RNA secondary structures and its application to RNA motif detection, BMC Bioinformat., 6:89.
10. M. Khaladkar, V. Bellofatto, J. T. L. Wang, B. Tian, and B. A. Shapiro (2007), RADAR: a web server for RNA data analysis and research, Nucleic Acids Res., 35:W300–W304.
11. M. Khaladkar, V. Patel, V. Bellofatto, J. Wilusz, and J. T. L. Wang (2008), Detecting conserved secondary structures in RNA molecules using constrained structural alignment, Comput. Biol. Chem., 32:264–272.
12. I. L. Hofacker (2003), Vienna RNA secondary structure server, Nucleic Acids Res., 31:3429–3431.
13. M. Khaladkar, J. Liu, D. Wen, J. T. L. Wang, and B. Tian (2008), Mining small RNA structure elements in untranslated regions of human and mouse mRNAs using structure-based alignment, BMC Genomics, 9:189.
14. A. R. Gruber, R. Lorenz, S. H. Bernhart, R. Neubock, and I. L. Hofacker (2008), The Vienna RNA websuite, Nucleic Acids Res., 36:W70–W74.
15. H. M. Berman, W. K. Olson, D. L. Beveridge, J. Westbrook, A. Gelbin, T. Demeny, S.-H. Hsieh, A. R. Srinivasan, and B. Schneider (1992), The Nucleic Acid Database: a comprehensive relational database of three-dimensional structures of nucleic acids, Biophys. J., 63:751–759.
16. E. Bindewald, R. Hayes, Y. G. Yingling, W. Kasprzak, and B. A. Shapiro (2008), RNAJunction: a database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign, Nucleic Acids Res., 36:D392–D397.