
Chapter 10
Reactive Crystallization
10.1 INTRODUCTION
When supersaturation of a crystallizing compound is created by its formation between two chemical species via chemical reaction, ionization, or hydrogen bonding, the operation is termed reactive crystallization. These operations are also known as precipitation. The term reactive crystallization is generally applied only when the product is crystalline. The products of the more general term precipitation may be amorphous or crystalline.
The reaction may be between two complex organic compounds or may be neutralization by an acid or a base to form a salt of a complex compound or may be formation of co‐crystals between the compound and co‐crystal former via hydrogen bonding. These reactions can be very fast compared to both the mass transfer rates of the crystals and the growth rate of the crystals, possibly leading to high local supersaturations and, therefore, extensive nucleation.
Although crystallization by anti‐solvent addition shares many characteristics with that caused by chemical reaction, the processes often differ in the rate of creation of supersaturation (e.g. a rapid reaction leading to a compound of very low solubility). Reactive crystallization is also subject to other kinetic considerations which are sometimes less predictable than the known solubility effects caused by addition of an anti‐solvent.
In some cases, the fine crystals or precipitates resulting from high supersaturation (often in the range from 0.1 to 10 μm) are desired in order to meet specific needs for downstream processing or formulation. In most cases, however, these fine particles are not desired since they can be very difficult to handle in downstream processing—notably filtration, washing, and drying. The reader will note many uses of qualitative terms to predict the behavior of these complex systems. As in the entire field of crystallization, these wide brackets around possibilities (e.g. will it crystallize, will it form an oil first, will it stay amorphous, will it grow, will it nucleate, what is good mixing, what is low supersaturation, etc.?) are necessary because of the extreme species and conditions dependency of the crystallization of organic molecules. The guidelines offered are intended as such and, in addition, to provide a framework for experimentation to determine where a particular system may fit in the wide scope of crystallization behavior possibilities.
10.1.1 Utilization
This method of crystallization has become increasingly common in the pharmaceutical industry because organic molecules often have poor water solubility and must be converted to a salt form and a co‐crystal to improve water solubility and enhance bioavailability. Reactive crystallization/precipitation is also used in the fine chemicals industry to create fine particles for a wide variety of applications including photographic chemicals, dyes, printing inks, agrochemicals, topical formulations, and cosmetics.
The ultimate particle size distribution (PSD) from crystallization is dictated by the balance between nucleation and crystal growth rates. The processes indicated above, because of very high supersaturation, often result in rapid nucleation of too many particles and smaller than desired final product. The rapid nucleation and growth occurring may also result in impurity and/or solvent occlusion.
In addition, these normally small particles may agglomerate into weak structures which are readily broken up during aging in the crystallizer, transfer to a filter, on the filter, and/or in the dryer. The resulting low filtration rates, high solvent retention, poor washing, caking, and slow drying may be impractical for a production operation. Viscosities of the product slurries may be high because of high solids contents and poor two‐dimensional crystals. In addition, other properties of the bulk active product such as bulk density (high bulk), PSD (excessively fine, broad, and/or bimodal), and caking may be unsuitable for pharmaceutical formulation operations.
Intermediates in a chemical synthesis may also be produced by reactive crystallization and are also subject to impurity occlusion and poor downstream performance.
10.1.2 Literature
The literature contains some excellent discussions of the theory and practice of reactive crystallization. In particular, the reader is referred to the definitive book on this topic by Sohnel and Garside (1992), which contains both theoretical development of the key factors involved in precipitation and a summary of practical aspects.
In addition, excellent treatment of this topic may be found in the books by Mullin (2001) and Mersmann (2001). The major part of this literature is focused on the study of precipitation of inorganic compounds by reactions between ions. Specialized studies of organic compounds are also treated including, for example, ethylenediaminetetraacetic acid (EDTA) (Myerson 2002) and calcium oxalate (Marcant and David 1991). Guo et al. (2021) provides a nice review and discussion on co‐crystallization preparation, including reactive crystallization, together with other technique.
Mixing issues and chemical reactions are the subject of definitive books including Baldyga and Bourne (1999) and Zlokarnik (2001). In addition, several studies have appeared concerning theoretical and experimental studies on the effect of mixing on precipitation. These include Manth et al. (1996), Houcine et al. (1997), Torbacke and Rasmuson (2001), Åslund and Rasmuson (1992), and Phillips et al. (1999).
In their chapter in Mersmann (2001), Klein and David summarize the difficulties in scale‐up as follows: “The classical agitated industrial vessel seems to be too complicated in terms of turbulence or macro‐ and micromixing to allow a reliable scale‐up from laboratory experiments” (p. 557).
These scale‐up issues present some of the more difficult challenges in crystallization processes, as they do for other engineering operations. In most cases, the products obtained may be handled but with increased capital and operating expense for the cumbersome downstream operations. These costs may exceed those of the other steps in a manufacturing operation. In extreme cases, the products may be unsuitable for commercial use because of the poor physical properties discussed above.
This chapter outlines some guidelines that may be helpful in some cases in developing process options that are capable of improving the chemical purity and/or physical properties of the product of a reactive crystallization. These guidelines are discussed in the text to follow and illustrated in Example s 10.1 and 10.2. It is recognized that there are systems for which these alternative process options will not be successful in increasing particle size or improving purity. The system characteristics that cause the most difficulty are short nucleation induction time, rapid nucleation rate, and slow growth rate. The last of these can cause the most difficulty and, in the extreme, some compounds in which nucleation represents a substantial fraction of the release of supersaturation do not grow beyond the nucleation/growth phase. However, it is recommended that a development program include investigation of the options before concluding that no improvement in the basic particle obtained from a reaction can be realized. Guidelines for development are presented.
10.2 CONTROL OF PARTICLE SIZE
Because the rate of creation of supersaturation in reactive crystallization is dependent on the reaction kinetics, control of particle size can be difficult because slowing down the reaction is often difficult or undesirable. For compounds with fast crystallization kinetics, traditional methods such as reduced concentration and temperature may help, but the range of improvement may not be significant.
The rate of addition of the reagents, however, does provide a means to control supersaturation globally in the reactor but not locally since the reaction may be complete near the point of addition. Successful operation depends, therefore, on a careful balance between addition rate of the reagent(s), local supersaturation, global supersaturation, mass transfer, and crystal growth surface area. For compounds with slow crystallization kinetics, the rate of addition of reagents provides an effective means to control supersaturation.
Three possible courses of a reactive crystallization are illustrated in Figure 10.1. The equilibrium solubility line is essentially horizontal since most reactive crystallizations result in products that have low solubility in the solvent system and, therefore, may have small changes in solubility based on temperature. Because of the high supersaturations generated, primary nucleation is usually dominant, producing a large number of crystals. Three reagent addition strategies are illustrated; linear (A, B, C), programmed (A, D), and programmed with seed (A, E). It should be noted that the solubility at a supersaturation of 1.0 will normally be very low and the first amount of reagent added will create relatively high local supersaturation.
The differences in crystallization conditions for these three additions are shown by the degree of the addition that occurs above the metastable region, thereby producing excessive nucleation in the unseeded cases. This excessive nucleation may be avoided by addition with a sufficient amount and size of seed to maintain supersaturation within the metastable region. This strategy is discussed in Section 10.3.5.
Controlled supersaturation at the initiation of the addition of the reagent(s) requires an initial charge of seed to minimize uncontrolled nucleation and the resulting creation of an excess number of particles. It is desirable to develop the seed in a separate operation because it is difficult to ensure a robust process with a reproducible seed level using only the intrinsic reaction. This issue and methods are discussed in Section 10.3.4.

Figure 10.1 Schematic representation of addition modes for reagents in reactive crystallization.
10.2.1 Controlling for Growth
The objective of the remainder of this chapter is to suggest methods of achieving growth dominated processes of organic compounds by reactive crystallization. Two examples of controlled processes are presented (Examples 10.1 and 10.2). In these cases, successful scale‐up was achieved by minimizing nucleation and promoting growth, thereby helping to minimize the scale‐up issues. As also indicated, this is not to suggest that all reactive crystallization processes will be responsive to the same methods since this approach requires that the compound in question has reasonable inherent growth rates. In the case of amorphous products, these process options are not expected to be of value because predictable and consistent solid phase growth rates are very difficult to obtain with amorphous solids. A possible exception is “growth” by agglomeration which is controlled by other factors. Reliance on agglomeration may be feasible in some cases and is discussed in Section 10.3.8.
The creation of fine particles may be desired for specific down‐stream processing or formulation requirements. Methods for control of these applications are also discussed in Section 10.5.
The complex issues regarding low solubility and the nucleation characteristics of induction time and nucleation rate must be minimized by the maintenance of low supersaturation so that growth can predominate. Methods for promoting this balance will be indicated. Prior to that discussion, the issues encountered in the development of a reactive crystallization process will be briefly reviewed.
10.3 KEY ISSUES IN ORGANIC REACTIVE CRYSTALLIZATION
Although the references cited above are focused primarily on inorganic precipitation, the key issues are essentially the same for organic compounds, with the exception that nucleation and growth can be slower in many cases. However, since many organic bases and acids are formed by reactive crystallization from their respective parent forms, the reactions can be very fast, leading to high supersaturation with potential for nucleation, as in inorganic precipitation.
As is elaborated in all of these discussions, the final physical attributes and possibly the chemical purity may be a function of several rate processes that are occurring both in series and in parallel. The most common procedure is to add a reagent to a solution of the organic compound to form the crystallizing species. The resulting rate processes include
- Mesomixing and macromixing of the added reagent to a degree sufficient to start a molecular‐scale contact and reaction.
- Micromixing to molecular‐scale homogeneity combined with mesomixing.
- The chemical reaction itself to produce the reaction product at low supersaturation.
- The formation of clusters of the reaction product in solution, ultimately exceeding the solubility.
- The creation of an insoluble phase consisting of the reaction product now as an amorphous “oil” or solid or crystalline solid—nucleation.
- Continuation of supersaturation as the reagent is added.
- Continuation of simultaneous nucleation and growth.
- Possible secondary changes including agglomeration, flocculation, crystal breakup by shear, change in crystal form, ripening.
- Completion of addition.
- Continuation of secondary changes.
Note: Precipitates that are initially an oil or amorphous can “turn over” and become crystalline at any point after formation of clusters or remain amorphous.
These many interacting rate processes may all be at play in a reactive crystallization. Successful development of a consistent and scalable process requires evaluation of the potential for each to be significant so that methods of control can be developed. Two additional factors that have potential for critical influences are
- Impurities—sometimes referred to as additives if they are intentionally introduced.
- Mixing both in a mixer configuration and at the addition point in a vessel.
The critical issues that must be addressed in experimental development and scale‐up studies are outlined below. Mixing issues are involved in all aspects and will be discussed first.
10.3.1 Mixing Issues
The extensive literature on the key mixing issues of macromixing, mesomixing, and micro‐mixing, for chemical reactions is applicable to mixing issues encountered in reactive crystallization. A major focus of this literature is on the effect of mixing on complex reactions with multiple products without separation of a second phase. The issues in reactive crystallization are usually confined to reactions with a single product that does separate as a second phase. The reader is referred to Chapter 5 in this book and the references cited therein for a review of this field of mixing. The following is a brief discussion of mixing issues in reactive crystallization.
10.3.1.1 Micromixing
Micromixing is a key factor in reactive crystallization because the time of blending to the molecular level is critical to both the chemical reaction and the induction time for nucleation. Micromixing times in stirred vessels are critically affected by
- location of the reagent(s) feed pipe(s)
- impeller speed
- impeller type
The range of micromixing time at various locations in a stirred vessel can vary by a factor of >20 at the same impeller speed and also by large factors for different speeds and types. This time is critical, depending on the reaction rate and the nucleation induction time, in determining the nucleate particle size and the complex sequence of events in the micro‐mixed zone. For fast reactions and short nucleation and induction times, a particle leaving this zone may already be a crystalline or an amorphous solid. For slower reactions and longer induction times, no phase change may occur in this zone.
A critical factor in micromixing and mesomixing in stirred tanks is the location of the reagent feed pipe(s). Experiments at different feed pipe locations with the reactive crystallization under investigation can readily determine whether micromixing and/or mesomixing issues are controlling. A change in mean particle size and PSD at different feed pipe locations would indicate mixing sensitivity. On the other hand, if no significant differences are observed, a different crystallization property of the system, such as nucleation induction time, may be slow relative to the micromixing time. The reaction itself could also be slow and occur in the bulk mixing regions of the vessel. Experimental work on this effect for barium sulfate is described by Nienow and Inoue (1993) and for calcium oxalate by Marcant and David (1991).
Organic compounds exhibit nucleation induction times over a wide range. Faster times are on the order of milliseconds that are in the same range as micromixing times in stirred tanks. Mixing effects can then be expected to be significant on particle formation. Reagent feed into poorly mixed regions can experience 10–100 times slower micromixing and mesomixing times, thereby making mixing issues more severe even with longer nucleation induction times. Reaction times can bracket these time frames, thereby adding to the complexity of particle formation. These effects are illustrated by the Damkoehler number, as discussed in Chapter 5 and illustrated in Figure 5.2.
Micromixing times can be reduced by an order of magnitude by in‐line mixers such as rotor–stator homogenizer, or impinging jets, as discussed in Chapters 5, 6, and 9. These devices can be utilized effectively in reactive crystallization and are discussed by Condon (2001) and Johnson (2003), in journal articles by Johnson and Prud’homme (2003) and Hacherl (Condon) (2003), and in Section 10.5.
10.3.1.2 Macromixing
Macromixing is another term for bulk blending in stirred vessels. Compounds with longer nucleation induction times and/or reagents with slower reaction rates may not form particles in the micromixing or mesomixing zones but will eventually do so in the bulk blending phase of the overall mixing process. Although these are reactive crystallizations, they will be influenced more by antisolvent crystallization effects of mixing.
10.3.1.3 Mesomixing
Mesomixing effects occur in stirred vessels when the reagent(s) feed rate is faster than the macromixing rate, resulting in a plume of reagent concentration that is not yet mixed uniformly throughout the vessel to the molecular level. This mixing effect is important on scale‐up and results in the need for longer reagent addition times on the larger scale to achieve the same reaction selectivity as on a smaller scale (Baldyga and Bourne 1999, pp. 732–744). Its effect on reactive crystallization would be expected to require increased addition time to achieve an equivalent mean particle size and PSD, although data on this dependency are difficult to document because of the many interacting variables.
10.3.2 Mixing and Growth
Mixing effects are also critical in reactive crystallizations designed to minimize nucleation and maximize crystal growth. This design strategy is discussed in Section 10.3.6. The issue here is to achieve micromixing of two reagents faster than the nucleation induction time and reaction time as well as effective meso‐ and macromixing in order to create supersaturation at a controlled rate. Under these conditions and in the presence of sufficient seed surface area and mass transfer to the growing crystals, nucleation can be minimized in favor of growth. However, excess mixing could cause a decrease in induction time and an increase in nucleation rate as well as crystal fracture by shear, thereby requiring a balance in power input.
These contrasting mixing requirements form the basis of the mixing dilemma that is particularly applicable to reaction crystallization. It is apparent that each reactive system will respond differently because of the extreme ranges in organic compound inherent characteristics including reaction rate, induction time, nucleation rate, growth rate, and shear sensitivity. These issues are discussed in the references cited above and in Chapters 5 and 6. Laboratory experimentation, coupled with numerical simulation, is highly desirable to find the balance required. Scale‐up studies are then necessary to ensure that these balances can be maintained at the predicted conditions. Examples of a growth‐dominated reactive crystallization are presented (Examples 10.1 and 10.2).
10.3.3 Induction Time and Nucleation
Induction time, nucleation rate, and nucleate particle size all can play key roles in determining the course of a reactive crystallization. These issues are discussed in the crystallization literature and in Chapters 2, 4, and 6 in this book for general crystallization, and their importance in reactive crystallization will be summarized here.
Induction time is a key issue because of its relationship to reaction time and mixing time. At the point of reagent introduction, as outlined above, mixing, reaction, and nucleation may be occurring within the same time frame more or less in parallel or may occur in series, depending on the respective time constants. If they occur in parallel, mixing may not be complete to the molecular level before reaction starts. If induction time is also short, incorporation and/or entrapment of unreacted substrate in a nucleating mass in regions of local high supersaturation are possible. In this case,
Ideally, the mixing is sufficiently intense locally to achieve reagent blending to the molecular level before reaction, and the reaction is complete in less than the induction time so that nuclei can form from the expected molecular composition. In this case,
Mixing time can be varied as discussed above, but reaction and induction time are both system‐specific. Reaction time and induction time are both concentration dependent, and induction time also is a function of supersaturation (tind decreases with increasing S).
Also because of high supersaturation, nucleation may be primarily homogeneous rather than secondary, although this is also system dependent and may be reversed for organic molecules. Nucleation rate is a key issue because of its impact on the number of crystals formed.
These interacting factors may be uncontrollable without control of local conditions to prevent extensive nucleation.
10.3.4 Supersaturation Control
Control of both local (point of addition) and global supersaturation is essential, as in all crystallizations, if a satisfactory balance between nucleation and growth is to be achieved. This is particularly relevant to reactive crystallization because of the creation of local high supersaturation of these low‐solubility compounds that is unavoidable at the point of reaction. In addition to the mixing issues outlined above, the critical variables in minimizing supersaturation are as follows:
- Addition time of the reagent—must be long enough to maintain the global supersaturation in the metastable region.
- Sufficient initial seed area—to avoid or minimize nucleation at the outset of addition and prevent formation of an excessive number of nuclei, which would limit overall growth.
- Continuing balance between addition rate and growth surface area—to promote growth and avoid continuing nucleation, which would result in a bimodal distribution.
These issues are shown schematically in Figures 10.1 and 10.2. Figure 10.1 is discussed in Section 10.2. Figure 10.2 shows a similar relationship, and the region of concentration in the metastable zone to be maintained throughout the addition is indicated.
10.3.5 Seeding
Seeding is the key to achieving control of a reactive crystallization process. Without seeding, excessive nucleation can be expected in most systems, resulting in severe limitation on the final crystal size by creating an excessive number of particles. The guidelines outlined below have been found to be useful in several cases and will be further illustrated in Examples 10.1 and 10.2. For low growth rate compounds, however, growth of suitable seed material and significant growth of crystals during reagent addition may not be realized. In this case, a growth‐dominant process may not be achievable.
- The number, surface area, and surface condition of seed crystals are critical to successful minimization of nucleation and realization of growth. (Seeding issues are also discussed in Chapter 6, and only those issues of primary consideration in reactive crystallization are discussed here.)

Figure 10.2 Schematic representation of reagent addition time (overall time for a run) and the metastable region indicating the recommended region for promoting growth and minimizing nucleation.
- The recommended seed mean particle size is approximately one‐half of the expected final mean particle size.
- The recommended seed amount is at least of 10–15% (the amount necessary to maximize growth by providing sufficient initial surface area to minimize nucleation).
- The recommended method is to generate wet seed in situ, add seed in a slurry or as a heel from a previous batch. Dry seed or dry‐milled seed are not recommended. Wet milling or sonication, which can reduce the particle aspect ratio, is recommended between batches to maintain the desired seed size as necessary.
- Recommended seed preparation is by recrystallization from a suitable solvent(s). For in situ wet seed/particle generation, additional heat/cool cycles to improve aspect ratio is recommended (Initial preparation by reactive crystallization without seed will typically give seeds that are too small for successive use.)
Note: Seed growth by heat/cool temperature cycling is an effective means of growing seeds for initial use. This method is also useful in determining the potential for a particular compound to grow. This method is discussed in Chapter 6 and in Example 10.1.
The requirement for increased amounts of seed for reactive crystallization compared with the cooling, evaporation, and antisolvent methods is discussed by Mullin (2001, p. 339). Amounts of seed up to 50% are indicated to be necessary in recycle systems “to provide the seed area necessary.” The requirement for this increased amount is the direct result of the rapid development of supersaturation by reaction and the need to have sufficient surface area for growth throughout the operation, especially at the start of reagent addition.
10.3.6 Crystal Growth
The first issue with regard to growth is to determine if the compound in question has potential for growth. Growth potential may be very difficult to determine definitively because of (i) large dependence on impurities and (ii) lack of patience. Once established, growth can be used for initial seed preparation. Once seed is grown, all crystallizations are started with proper seeds.
Agglomeration and/or aggregation are common in reactive crystallization and should not be confused with true growth (see Section 10.3.8). Secondary growth phenomena can also be expected, such as growth rate dispersion and size‐dependent growth (see Section 10.3.8).
10.3.6.1 Additional Strategies for Growth
To increase the probability of achieving and maintaining a primary growth process, the entire operation must be maintained in the metastable region, as shown schematically in Figures 10.1 and 10.2. The three critical factors that can achieve this condition are (i) mixing to minimize local supersaturation at the point of addition, (ii) limiting the addition rate to prevent buildup of a bulk concentration of the reaction product, and (iii) sufficient seed surface area. The rates of mass transport through the film surrounding each crystal and the incorporation into the crystal lattice by surface integration reach a balance so that they are essentially equal. The key to successful operation is to maintain the bulk concentration sufficiently low to allow the rate of surface integration to control so that transport through the film does not create a region of high concentration at the surface that could result in local nucleation. The effects of addition time and seed area on growth conditions are illustrated in Figure 10.2.
Since the inherent growth rate of many organic compounds is relatively slow, addition times may be long in order to achieve supersaturation control within the metastable zone. Higher addition rates can result in nucleation and the creation of a bimodal distribution. Experimentation to determine acceptable addition rates can be evaluated by focused beam reflectance measurement (FBRM) and other in situ, online methods (Chapter 2) or microscopic observation of the crystal slurry which could reveal the presence of fines. These issues are highlighted in the examples.
An addition strategy, termed programmed feed concentration, has been proposed by Lindberg and Rasmuson (2000) in which the initial feed concentration of the reagent is kept low and then increased as the addition proceeds. This strategy is analogous to programmed cooling (Chapter 7) or to programmed addition of anti‐solvent (Chapter 9). This programmed addition strategy is a natural choice for in situ wet seed/particle generation with in‐line mixer, where reagents can be added directly into the in‐line mixer with controlled and scalable mixing environment.
By limiting the reagent concentration at the most critical stage, the resulting mean particle size can be increased.
A method of determining the maximum addition rate consistent with avoiding nucleation is to experiment with increasing feed rates with all other variables held constant. The addition rate at which nucleation is observed, as indicated by the appearance of a bimodal distribution, is then the limiting addition rate. In situ methods of making this determination are valuable in this experimentation, as discussed in Chapter 2.
10.3.7 Impurities/Additives
The large dependence of growth rate on the presence or absence of impurities is well known and can be critical to the success of achieving growth of seed and product. Growth can be totally inhibited by levels of impurities as low as 0.1% but be satisfactory at >50%. This extreme variation is a function of the molecular structure of the impurity and its ability to retard or block surface incorporation. Excellent discussions of these issues may be found in the referenced texts. Sohnel and Garside (1992, p. 159) note that despite significant study, “no fully reliable general theory that does not need experimental verification of each case has so far been worked out to allow prediction of the influence of a certain impurity on the behavior of a given system or of the concentration at which the admixture becomes effective.” A case of dissolved impurities causing significant decreases in growth—including stopped growth under supersaturated conditions—may be found in Example 13.6.
Initial attempts to establish growth potential must be accompanied by careful evaluation of impurity contamination. This can be difficult when the structure of the impurities may not be known. One experimental technique to evaluate the effect of impurities that are inherent in the process but whose structures may not be known is to recrystallize the subject compound both from pure solvents and from the same solvents “spiked” with mother liquors from a primary crystallization. This technique can be very successful in revealing whether crystal growth is affected by the impurities in the spiked liquors.
In any event, extensive purification in the laboratory by any applicable method, including preparative high‐performance liquid chromatography, is recommended as part of growth potential determination studies. The importance of this effort cannot be over‐stated since a particular compound may not grow only because of the presence of impurities and not because of inherent crystal lattice incorporation restrictions.
The use of additives to influence crystal habit has received much experimental and theoretical attention. However, it is not discussed here because these techniques are not generally utilized in the pharmaceutical industry due to limitations on the addition of additives.
10.3.8 Secondary Effects
10.3.8.1 Oiling Out, Amorphous Solids, Nucleation, Agglomeration, and Growth
Reactive crystallization operations are subject to oiling out and/or agglomeration because of the inherently high local supersaturations encountered. As indicated in Section 10.3, the formation of a crystal may be preceded by oiling out as the first physical form that may or may not be observed (see also Chapter 6, Section 6.4). This oil may separate as a second phase because of the normally extremely low solubilities of the reaction products that result from the chemical reaction. This low solubility can cause a second liquid phase to form on a time scale that is shorter than the nucleation induction time. These issues are considered in Ostwald’s Rule of Stages.
The next event could be solidification of the oil into an amorphous solid or direct nucleation to a crystalline form. In either case, the fine particles are subject to agglomeration as the particles collide and stick together because of the oily/sticky nature of the solid surfaces at this point. Assuming a crystalline form results from this series of events, these nuclei can then grow to some extent, as determined by the system‐specific characteristics of the compound. This process continues throughout the addition of the reagent(s).
The final mean particle size and PSD will be determined by the number of nuclei created by these steps. If nucleation continues throughout the addition, the mean particle size will be small, perhaps <10 μm, and the PSD will be broad and possibly bimodal because of the large number of nuclei produced and the conditions allowing some crystals to grow while nucleation is creating small crystals.
If, on the other hand, there is a sufficient quantity of seed present initially and if the reagent(s) is added at a sufficiently slow rate with sufficiently good mixing, the low supersaturation created could be well below the metastable limit, thereby reducing or eliminating nucleation and supporting growth on the seed crystals. As indicated above, 10–20% of seed crystals could then be expected to approximately double in size, depending on the aspect ratio of the crystal.
The oiling out and agglomeration possibilities can complicate or prevent successful crystallization. In an extreme case of oiling out, a solid phase never forms and the oil drops could coalesce or remain dispersed. This condition could result from several factors, including a high impurity level inhibiting nucleation, a low melting point solid, and further depression of the melting point by mutual solubility of the solid and a component of the solvent mixture.
Agglomeration can complicate an operation by forming large structures of crystals or amorphous solids that can trap impurities, unreacted reagents, and/or solvents. These structures may be strong enough to stay coalesced and persist through filtration, washing, and drying. In some cases, agglomeration can be beneficial to these downstream operations if the trapping of impurities is not excessive.
In many cases, however, agglomerates are weakly structured and break down on continued mixing, during pumping, during filtration, and/or during drying. This disintegration could be difficult to handle in these downstream operations, and primarily for this and impurity entrapment issues, agglomeration should be avoided when possible.
Methods to avoid oiling out and agglomeration are essentially the same as those described above for favoring growth. The single most important consideration is avoiding high local supersaturation to the greatest extent possible by good mixing, seeding, and slow addition.
10.3.8.2 Growth Rate Dispersion and Size‐Dependent Growth
Other secondary effects, which are not exclusive to reactive crystallization, are size dependent growth and growth rate dispersion. These effects may not be separable, but both can change the final mean particle size and PSD. Both are discussed in Sohnel and Garside (1992, pp. 103–105) and in Chapter 4 of this book.
10.3.8.3 Reactive Crystallization to Increase Reaction Selectivity
The selectivity of a consecutive chemical reaction may be increased above that predicted from its rate constant ratio if the reaction is run under conditions in which the product crystallizes while the reaction is in progress. Crystallization of the product reduces its concentration in solution, thereby making it less available for overreaction. The topic of increasing reaction selectivity in the presence of, or by the creation of, a second phase (e.g. by crystallization), has been discussed by Sharma (1988) and Paul et al. (2003, chapter 13). This important application of crystallization is illustrated in Examples 13.2 and 13.3.
10.4 CREATION OF FINE PARTICLES—IN‐LINE REACTIVE CRYSTALLIZATION
In‐line methods are applicable to reactive crystallization with systems with a relatively fast reaction rate and a short nucleation induction time. There are different in‐line devices that have potential for these applications including impinging jets, vortex mixers, and rotor–stator homogenizer. The reader is referred to Chapter 9 for a discussion of in‐line mixers as applied to antisolvent crystallization. The application to reactive crystallization is similar—creation of high supersaturation in a short time. For impinging jet mixer, one limitation is in the range of flow rate ratios of the reacting feed streams to balance the momentum of impact. Although some adjustments in feed tube diameter can be helpful in maintaining the momentum balance by equalizing velocities, the ratio of feed streams may be limited to 80/20. This limit may be an issue in reactive crystallization applications because the feed streams may have volume ratios of 90/10 or more. This would be less a constraint for rotor–stator type mixer where mixing is achieved via the rotor–stator design and independently controlled rotor rotating at a tip speed of tens of meter/sec.
Impinging jets have been studied experimentally for the reactive crystallization of calcium oxalate by Condon (2001) and Hacherl (Condon) (2003). The primary objective of this study was the determination of the capability of the jet system to produce fine particles. Particles with a mean particle size of 2 μm and a PSD of 1–3 μm were obtained. In addition, the effect of jet velocity on mean particle size and PSD was studied, and a correlation in the expected direction of smaller particle size with increased velocity was obtained. The flowsheet is shown in Figure 10.3.
This system is complicated by the appearance of two hydrate forms, mono‐ and dihydrate. The dihydrate is favored at higher excess oxalate local concentrations, while the monohydrate is favored at higher excess calcium local concentrations. Local concentrations within the device were shown to be possible and to cause a shift in hydrate ratio. The particle size results from two typical runs in this study at two jet velocities are shown in Figure 10.4. Increasing the jet velocities decreases the particle size. A photograph of the impingement plane is shown in Figure 10.5 from Condon (2001). The shift of vertical angle of impingement plane is clearly observed.
Liu (2015) applied a similar concept of impinging jet mixer for the crystallization of sodium cefuroxime. In the set‐up, the impinge jet mixer is directly built into a 1‐l crystallizer. Both acid and base streams are simultaneously fed into the impinging jet mixer at a flow rate ratio of ~1/4. In the meanwhile, the batch is continuously withdrawn from the bottom of crystallizer at a residence time close to ~500 seconds. As shown in Figure 10.6, the particle size distribution by the impinging jet approach is much more uniform and narrower in comparison to conventional stirred tank approach.
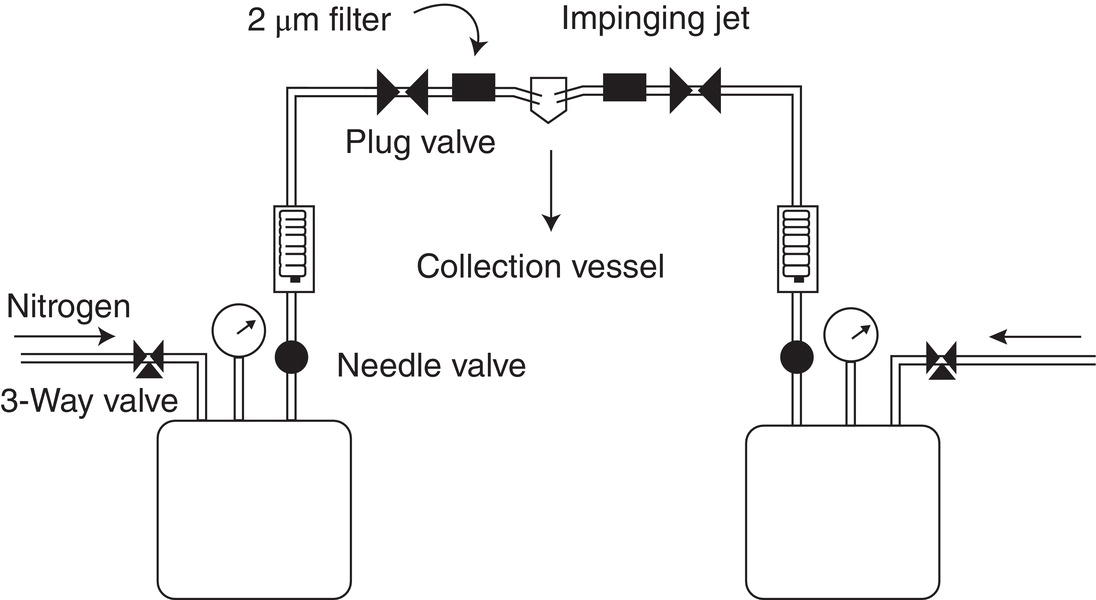
Figure 10.3 Impinging jet crystallization apparatus.
Source: Condon (2001), with permission Rutgers University.

Figure 10.4 Size distribution of calcium oxalate crystallized at (a) 2 and (b) 5.5 m/sec jet velocity.
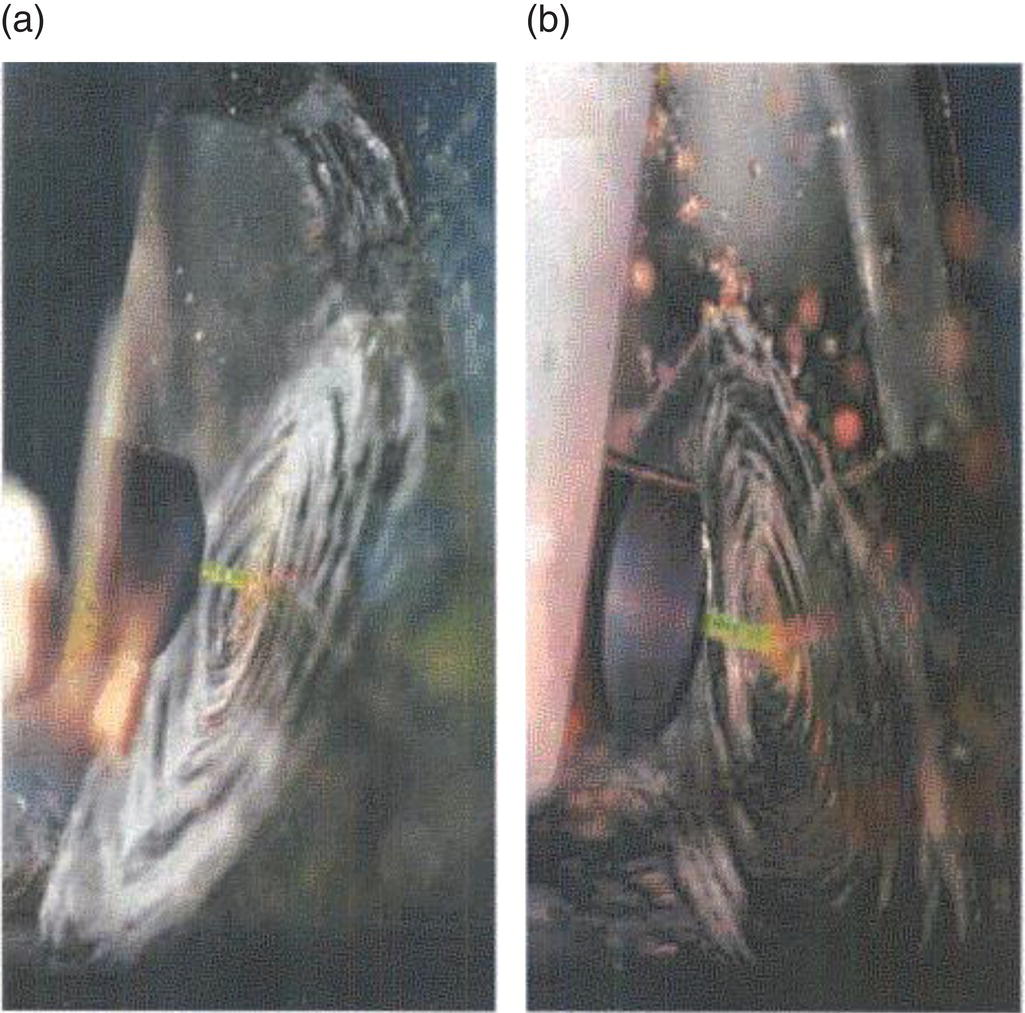
Figure 10.5 Photographs of impingement planes in a free jet (a) at feed velocities of 5.1 and 5.5 m/sec and (b) at 5.5 and 5.1 m/sec of calcium chloride and sodium oxalate, respectively.

Figure 10.6 Size distribution of sodium cefuroxime crystallized by conventional stirred tank (left) and impinging jet crystallizer (right).
Source: Liu (2015) with permission Elsevier.
This design is fully consistent with the concept of in situ wet seed/particle generation technique with in‐line mixer. Within the in‐line mixer, high supersaturation is generated under high mixing intensity which promotes particle nucleation. The generated nuclei acts as seed and is re‐circulated back to the crystallizer to grow into larger crystals under controlled supersaturation.
10.5 PROCESS DESIGN AND SCALE‐UP
Development and scale‐up of reactive crystallization/precipitation processes can present some of the most difficult challenges in the field. The reader is referred to the quotation in Section 10.1.2 above as a reminder of this difficulty.
However, the authors have participated in development and scale‐up of some successful reactive crystallization processes, and the examples to follow (Examples 10.1 and 10.2) are included to illustrate the concepts and application of the principles discussed above in these processes. These developments were based on the three essential concepts of seeding, control of supersaturation, and promotion of growth, as described above. The key variables are, therefore,
- amount of seed
- size of seed
- addition rate
- mixing intensity