
1
General
1.1 Definition: Pigments and Dyes
Colourants are classified as either pigments or dyes. Pigments are inorganic or organic, coloured, white or black materials that are practically insoluble in the medium in which they are incorporated. Dyes, unlike pigments, do dissolve during their application and in the process lose their crystal or particulate structure. It is thus by physical characteristics rather than by chemical composition that pigments are differentiated from dyes [1]. In fact, both are frequently similar as far as the basic chemical composition goes, and one structural skeleton may function either as a dye or as a pigment.
In many cases the general chemical structure of dyes and pigments is the same. The necessary insolubility for pigments can be achieved by avoiding solubilizing groups in the molecule or by forming insoluble organic structures. Carboxylic and especially sulfonic acid functional groups lend themselves to the formation of insoluble metal salts (lakes); the formation of metal complex compounds without solubilizing groups and finally suitable substitution may decrease the solubility of the parent structure (e.g. carbonamide groups).
Pigments of many classes may be practically insoluble in one particular medium, yet dissolve to some extent in another. Partial solubility of the pigment is a function of the application medium and processing conditions, especially of the processing temperature. Important application properties of pigments and/or pigmented systems, such as tinctorial strength, migration, recrystallization, heat stability, lightfastness, and weatherability, are often determined by the portion of pigment that dissolves to a minor degree in the vehicle in which it is applied.
Monohydrazone yellow pigments of the Hansa Yellow type (e.g. Pigment Yellow (P.Y.) 1, P.Y.3; Section 2.3.4.2) may serve as an example. Their solubility in air-dried alkyd resin systems is so negligible that they are considered insoluble, which explains their frequent use in such media. Since their solubility increases with increasing temperature, they migrate considerably in vehicles such as various oven-dried varnish systems or in plastics. This results in bleeding or blooming (Section 1.6.3). Strong migratory tendencies preclude their use in such high temperature applications. Even slight temperature changes in the course of pigment incorporation into its application medium may often determine the commercial fate of a pigment. Moreover, the inherent tinctorial properties of a product in a particular vehicle system are sometimes compromised by difficulties such as recrystallization, which arises through a certain solubility of the pigment in its medium.
Under certain circumstances, it may even be advantageous to have a pigment dissolved to some degree in its binder system in order to improve certain application properties such as tinctorial strength and rheological behaviour. Such conditions arise when special amine-treated diarylide yellow pigments are incorporated in toluene-based publication gravure inks (Section 1.8.1.1). In toluene, up to 5% of the amine-treated pigment may be either dissolved or dispersed to a nearly molecular level. This improves the tinctorial strength and decreases the viscosity, which in turn enhances the rheology of the pigmented ink. The performance of a colourant in its role as a commercial pigment is therefore defined by its interaction with the application medium under the conditions that govern its application.
1.1.1 Organic and Inorganic Pigments
In some application areas, inorganic pigments are also used to an appreciable extent, frequently in combination with organic pigments. A comparison of the respective application properties of inorganic versus organic pigments shows some fundamentally important differences between the two families.
Most inorganic pigments are extremely weatherfast (Section 1.6.6) and many exhibit excellent hiding power (Section 1.6.1.6). Their rheology is usually an advantage (Section 1.6.8), being superior to that of most organic pigments under comparable conditions. In white reductions, however, many inorganic pigments have much less strength than organic pigments. With few exceptions such as Bismute Vanadate, Molybdate Red, Chrome Yellow, and cadmium-based pigments, inorganic pigments provide dull shades. Since there are only relatively few inorganic types, the spectral range that is accessible by inorganic pigments alone is very limited. Many hues cannot be produced in this manner.
Inorganic pigments not only exhibit colouristic limitations but also frequently present application problems. Ultramarine Blue, for instance, is not fast to acid, while Prussian Blue must not be exposed to alkalis. Such limitations preclude the application of, especially, Prussian Blue in paints that are to be applied to a basic substrate (e.g. exterior house paints). In the red range of the spectrum, iron oxide red pigments produce weak hues of comparatively little brilliance. Molybdate Reds and Chrome Yellows lend themselves to a host of applications but are nevertheless sensitive to acids and light. There are stabilized versions of such pigments that claim improved lightfastness and acid resistance. These products also claim to be chemically fast to hydrogen sulfide, which affects the brightness of a coating through sulfide formation. However, if the particle surfaces of such types are damaged during the dispersion process, the above-mentioned deficiencies are apparent at the damaged site.
Poor tinctorial strength and lack of brilliance restrict the use of inorganic pigments in printing inks. There are areas of application, however, where it is hardly, if at all, possible to replace the inorganic species by an organic pigment. The ceramics industry, for example, requires extreme heat stability, which precludes the use of organic compounds. Thus, the organic and inorganic classes of pigments are generally considered complementary rather than competitive.
1.2 Historical
The history of pigment application dates back to prehistoric cave paintings, which give evidence of the use of ochre, haematite, brown iron ore, and other mineral-based pigments more than 30 000 years ago. Cinnabar, azurite, malachite, and lapis lazuli have been traced back to the third millenium BC in China and Egypt. With Prussian Blue, in 1704 the first non-natural inorganic pigment was synthesized. It was not until a century later that Thenard produced his Cobalt Blue. Ever increasing expertise and technology led to the production of Chrome Yellow, Cadmium Yellow, several synthetic iron oxides covering parts of the ranges of yellow, red, and black hues, Chrome Oxide Green, and Ultramarine.
Important twentieth-century developments include the addition of Molybdate Red to the series of inorganic synthetic colouring matters in 1936; Titan Yellow followed in 1960.
Later newly developed inorganic pigments have been introduced to the market, such as bismuth–molybdenum–vanadium oxide pigments for lead-free formulation, or cerium sulfide pigments, which can be used as a replacement for cadmium sulfide pigments.
The beginning of organic pigment application dates to antiquity. It is certain that the art of using plant and animal ‘pigments’ to extend the spectral range of available inorganic colourants by a selection of more brillant shades had been practised thousands of years ago. However, for solubility reasons, most of these organic colours would now be classified as dyes rather than pigments. Even in antiquity, they were used not only for dyeing textiles but also, due to their ability to adsorb on mineral-based substrate such as chalk and china clay, for solvent resistant coatings for decorative purposes. These materials later came to be known as lakes or toners. For thousands of years, derivatives of the flavone and anthraquinone series have been the major source of natural colours for such applications.
The beginning of the era of scientific chemistry was marked by the synthesis of large numbers of dyes for textile related purposes. Some of these were also applied to inorganic substrates by adsorption, for use as pigment toners. The commercially available soluble sodium salts of acid dyes were rendered insoluble, an essential property of pigments, by reacting them with the water-soluble salts of calcium, barium, or lead to form lakes. Basic dyes (commercially available as chlorides or as other water-soluble salts), on the other hand, were treated with tannin or antimony potassium tartrate to yield insoluble colourants, that is, pigments. Some of the early commercially important lakes, such as Lake Red C (Pigment Red 53:1) and Lithol Rubine (Pigment Red 57:1), released on to the market in 1902 and 1903, respectively, are still commercially important products (Section 2.7).
Entering the market in the late nineteenth century were the first water insoluble pigments that did not contain acidic or basic groups, namely, the red β-naphthol pigments (Para Red P.R.1, 1885). Falling into the same chemical class are Toluidine Red (P.R.3, 1905) and Dinitroaniline Orange (P.O.5, 1907), two members of this class of pigments that still enjoy commercial importance today. In 1909, Hansa Yellow (P.Y.1) was introduced to the market as the first monohydrazone yellow pigment. The first red Naphthol AS pigments followed in 1912, and the first commercial pioneers of the diarylide yellow pigment range, some of which had been patented as early as 1911 [2], appeared in 1935. Phthalocyanine blue pigments also appeared in 1935, followed by phthalocyanine green pigments a couple of years later [3]. The rapid advances in pigment chemistry led to such important classes of pigments as dihydrazone condensation pigments in 1954, quinacridones in 1955, dihydrazone pigments of the benzimidazolone series in 1960, the isoindolinone pigments in 1964 [4], and the diketopyrrolo-pyrrole pigments in 1986.
1.3 Classification of Organic Pigments
Several classification systems for organic pigments have been proposed over the years. Basically, it seems appropriate to adopt a classification system by grouping pigments either by chemical constitution or by colouristic properties. A strict separation of the two classification systems is not very practical, because the categories tend to overlap; however, for the purposes of this book it is useful to list pigments according to their chemical constitution.
A rough distinction can be made between hydrazone and nonhydrazone pigments. Most nonhydrazone pigments have polycyclic ring systems and are therefore known as polycyclic pigments. The group of hydrazone pigments can be further subdivided according to structural characteristics, such as by the number of hydrazone groups or by the type of diazo or coupling component. Polycyclic pigments, on the other hand, may be identified by the number and the type of rings that constitute the aromatic structure. Furthermore, there is a smaller group of pigments that contains neither a hydrazone moiety nor a polycyclic ring system.
1.3.1 Hydrazone Pigments (Formerly Called Azo Pigments) (Chapter 2)
Hydrazone pigments have the hydrazone group (NHN) in common. Formerly, hydrazone pigments were called ‘azo pigments’, because they were believed to contain the azo group NN. However, all commercial ‘azo’ pigments do not contain an azo group, but a hydrazone group instead (see Chapter 2, Section 2.1 for details). Thus, the correct name is ‘hydrazone pigments’ instead of ‘azo pigments’, and this name is used throughout this book.
The synthesis of hydrazone pigments is economically attractive, because the standard sequence of diazonium salt formation and subsequent reaction with a wide choice of coupling components allows access to a wide range of products. The hydrazone pigments can be subdivided into monohydrazone and dihydrazone pigments.
1.3.1.1 Monohydrazone Yellow and Orange Pigments (Formerly Called Monoazo Yellow and Orange Pigments) (Section 2.3)
Monohydrazone yellow pigments that are obtained by coupling a diazonium salt with acetoacetic arylides as coupling components cover the spectral range between greenish and medium yellow; coupling with 1-arylpyrazolones-5 affords reddish yellow to orange shades.
All members of this pigment family share good lightfastness, combined with poor solvent and migration resistance. These properties define and limit their application. Monohydrazone yellow pigments are used extensively in air-dried alkyd resin and in emulsion paints, and certain inks used in flexo and screen printing. Other applications are in letterpress and offset inks, as well as in office articles.
1.3.1.2 Dihydrazone Pigments (Formerly Called Disazo Pigments) (Section 2.4)
There is a dual classification system based on differences in the starting materials. The first and most important group includes compounds whose synthesis involves the coupling of di- and tetra-substituted diaminodiphenyls as diazonium salts with acetoacetic arylides (diarylide yellows) or pyrazolones (dihydrazone pyrazolones) as coupling components. The second group, bisacetoacetic arylide pigments, are obtained by diazotization of aromatic amines, followed by coupling to bisacetoacetic arylides.
The colour potential of dihydrazone pigments covers the colour range from very greenish yellow to reddish yellow and orange and red. Most show poorer lightfastness and weatherfastness – but better solvent and migration fastness than monohydrazone yellow and orange pigments. Their main applications are in printing inks and plastics, and to a lesser extent in coatings and toners for laser printing.
1.3.1.3 β-Naphthol Pigments (Section 2.5)
β-Naphthol pigments provide colours in the range from orange to medium red. The typical coupling reaction with β-naphthol as a coupling component yields such well-known pigments as Toluidine Red and Dinitroaniline Orange. Their commercial application in paints requires good lightfastness. Solvent resistance, migration fastness and lightfastness are comparable to the monohydrazone yellow pigments.
1.3.1.4 Naphthol AS Pigments (Section 2.6)
These pigments are obtained by coupling substituted aryl diazonium salts with arylides of 2-hydroxy-3-naphthoic acid (2-hydroxy-3-naphthoic acid anilide = Naphthol AS). They provide a broad range of colours from yellowish and medium red to bordeaux, carmine, brown and violet; their solvent fastness and migration resistance are only marginal. Naphthol AS pigments are used mainly in printing inks and paints.
1.3.1.5 Hydrazone Pigment Lakes (Formerly Called Azo Pigment Lakes) (Section 2.7)
In Europe, pigments of this type are known as ‘toners’, but since this term is used differently elsewhere we refer to them as ‘lakes’ throughout this book, although a chemically correct description would be ‘salt type pigments’.
Historically, ‘lakes’ refered to the first type of synthetic organic pigments made from water-soluble dyes by precipitation onto alumina hydrate (aluminium hydroxide).
Laked pigments are formed by precipitating a monhydrazone compound that contains sulfo and/or carboxy groups. The coupling component in the reaction may vary: monohydrazone yellow pigment lakes are based on acetoacetic arylides or 1-arylpyrazolones-5 (Section 2.3.1.2); β-naphthol lakes are derived from 2-naphthol; BONA pigment lakes use 2-hydroxy-3-naphthoic acid (Beta-Oxy-Naphthoic Acid); and Naphthol AS pigment lakes contain anilides of 2-hydroxy-3-naphthoic acid as a coupling component. Lakes may also be prepared from naphthalenesulfonic acids.
Monohydrazone yellow pigment lakes show good migration fastness and heat stability, making them useful products for plastics. Lake Red C is one of the commercially significant β-naphthol lakes. Limited lightfastness, which ranks far behind the non-laked β-naphthol counterparts, along with a tendency to migrate largely restricts their use mainly to the printing inks field.
Most BONA lake pigments provide an extra site for salt formation. Apart from the usual substituents, the diazo components of almost all BONA lake pigments contain a sulfonic acid function. Two acid substituents are thus available to form insoluble salts, which is the form in which these pigments are commercially available. Metal cations such as calcium, strontium, barium, magnesium or manganese combine with the organic anion to produce shades between medium red and bluish red. Their use in printing inks exceeds their increasing use in plastics and paints.
The organic acid group of Naphthol AS pigment lakes is part of the diazo component; a second site for salt formation can be provided by the coupling component. The plastics industry is the main user of such lakes.
Naphthalenesulfonic acid lake pigments are based on naphthalenesulfonic acid as a coupling component; introduction of an additional SO3H function as part of the diazo component is possible.
1.3.1.6 Benzimidazolone Pigments (Section 2.8)
Benzimidazolone pigments feature the benzimidazolone structure, introduced as part of the coupling component. The pigments that are obtained by coupling to 5-acetoacetylaminobenzimidazolone cover the spectrum from greenish yellow to orange; 5-(2-hydroxy-3-naphthoylamino)benzimidazolone as a coupling component affords products that range from medium red to carmine, maroon, bordeaux and brown shades. Pigment performance, including lightfastness and weatherability, is generally excellent. Pigments that satisfy the specifications of the automobile industry are used to an appreciable extent in automotive finishes. Benzimidazolone pigments are also used extensively to colour plastics and high grade printing inks.
1.3.1.7 Dihydrazone Condensation Pigments (Formerly Called Disazo Condensation Pigments) (Section 2.9)
These pigments can formally be viewed as resulting from the condensation of two carboxylic monohydrazone components with one aromatic diamine. The resulting high molecular weight pigments show good solvent and migration resistance and generally provide good heat stability and lightfastness. Their main markets are in the plastics field and in spin dyeing. The spectral range of dihydrazone condensation pigments extends from greenish yellow to orange and bluish red or brown.
1.3.2 Polycyclic Pigments (Chapter 3)
Pigments with condensed aromatic or heterocyclic ring systems are known as polycyclic pigments. The numerous pigment classes that fall into this category do not reflect their actual commercial importance; only a few are produced in large volumes. Their chief characteristics are good light- and weatherfastness and good solvent and migration resistance, but, apart from the phthalocyanine pigments, they are generally also more costly than hydrazone pigments.
1.3.2.1 Phthalocyanine Pigments (Section 3.1)
Phthalocyanine pigments are derived from the phthalocyanine structure, a tetraaza-tetrabenzoporphine. Although this basic molecule can chelate with a large variety of metals under various coordination conditions, today only the copper(II) complexes are of practical importance as pigments. Excellent general chemical and physical properties, combined with good economy, make them the largest fraction of organic pigments in the market today. Copper phthalocyanine blue exists in several crystalline modifications. Commercial varieties include the reddish blue alpha form, as stabilized and nonstabilized pigments, the greenish blue beta modification and, as yet less important, the intense reddish blue epsilon modification. Bluish to yellowish shades of green pigments may be produced by introduction of chlorine or bromine atoms into the phthalocyanine molecule.
1.3.2.2 Quinacridone Pigments (Section 3.2)
The quinacridone structure is a linear system of five anellated rings. These pigments largely have the same performance attributes as phthalocyanine pigments. Outstanding light- and weatherfastness, resistance to solvents and migration resistance justify the somewhat higher market price in applications for high grade industrial coatings, such as automotive finishes, for plastics and special printing inks. Unsubstituted trans-quinacridone pigments are commercially available in a reddish violet beta and a red gamma crystal modification. One of the more important substituted pigments is the 2,9-dimethyl derivative, which affords a clean bluish red shade in combination with excellent fastness properties. Solid solutions of unsubstituted and differently substituted quinacridones and blends with quinacridone quinone resulting in reddish to yellowish orange pigments are commercially available; in contrast, 3,10-dichloroquinacridone as yet enjoys only limited success as a pigment.
1.3.2.3 Perylene and Perinone Pigments (Section 3.4)
Perylene pigments include the dianhydride and diimide of perylene tetracarboxylic acid along with derivatives of the diimide; while perinone pigments are derived from naphthalene tetracarboxylic acid.
Commercially available types provide good to excellent lightfastness and weatherability; some of them, however, darken upon weathering. A number of them have excellent heat stability, which renders them suitable for spin dyeing. They are also used to colour polyolefins that are processed at high temperatures. The list of applications includes high grade industrial coatings, such as automotive finishes, and, to a lesser degree, special printing inks for purposes such as metal decoration and poster printing.
1.3.2.4 Diketopyrrolopyrrole (DPP) Pigments (Section 3.5)
The basic skeleton of this group of pigments consists of two anellated five-membered rings each of which contains a carbonamide moiety in the ring.
This class of pigments presently has some commercially used representatives, one of them with great importance in the market. In full shades and white reductions, the pigments afford shades in the colour range from orange to medium and bluish reds. The pigments are used primarily in high grade industrial coatings, including automotive finishes and in plastics because of their excellent lightfastness and weatherfastness as well as their good heat stability.
1.3.2.5 Thioindigo Pigments (Section 3.6)
4,4′,7,7′-Tetrachlorothioindigo with a reddish violet shade reigns supreme as a pigment amongst the derivatives of this indigo. It can be used for bordeaux shades in automotive refinishes. Thioindigo pigments are generally used in industrial coatings and plastics for their good lightfastness and weatherfastness in deeper shades.
1.3.2.6 Pigments Derived from Anthraquinone (Section 3.7)
Apart from some nonclassified pigments such as Indanthrone Blue (P.Bl.60), the anthraquinone pigments, which are structurally or synthetically derived from the anthraquinone molecule, can be divided into the following four groups of polycyclic pigments.
1.3.2.6.1 Anthrapyrimidine Pigments
The commercially leading member of this class is Anthrapyrimidine Yellow, which in very light white reductions affords a greenish to medium yellow with excellent weatherfastness. It lends itself primarily to application in industrial coatings such as automotive metallic finishes or to modification of the shades of automotive finishes.
1.3.2.6.2 Flavanthrone Pigments
Flavanthrone Yellow, the only commercially used flavanthrone, is a moderately brilliant reddish yellow. Excellent lightfastness and weatherfastness, combined with good solvent and migration resistance, make this pigment an attractive supplement to Anthrapyrimidine Yellow, mainly in the automotive finish industry.
1.3.2.6.3 Pyranthrone Pigments
Commercial attention focuses on the derivatives of the pyranthrone molecule at a varying level of halogenation. Most are orange, but others exhibit a dull medium to bluish red shade. Owing to their good weatherfastness pyranthrone pigments are used for high grade industrial finishes.
1.3.2.6.4 Anthanthrone Pigments
Dibromoanthanthrone is the only commercial pigment within this group. Qualities such as outstanding light- and weatherfastness justify the relatively high cost for application in high grade industrial coatings such as automotive finishes. The transparent pigment provides shades of scarlet for metallic finishes.
1.3.2.7 Dioxazine Pigments (Section 3.8)
Dioxazine pigments are based on triphenodioxazine, a linear system of five anellated rings. Apart from Pigment Violet 37, the commercially most representative one is Pigment Violet 23, an extremely lightfast and weatherfast compound with good to excellent solvent and migration resistance. Applications include the pigmentation of coatings, plastics, printing inks, as well as spin dyeing. Apart from producing violet shades, the pigment also lends itself to the shading of phthalocyanine blue pigments in colourations, particularly in coatings. It is also used to tone the light yellowish shade of titanium dioxide in whites and in shading carbon blacks that have a brownish cast.
1.3.2.8 Quinophthalone Pigments (Section 3.9)
Quinophthalone pigments have a polycyclic structure derived from quinaldine and phthalic anhydride. A few members of this class have gained commercial recognition for their very good temperature resistance. The main markets for their mostly greenish yellow shades are in the plastics and coatings industries.
1.3.2.9 Isoindolinone and Isoindoline Pigments
Although of comparatively good light- and weatherfastness and solvent and migration resistance, only a few members of the isoindolinone and isoindoline families are commercially available as pigments. Chemically classified as heterocyclic azomethines, these pigments produce greenish to reddish yellow hues. Isoindolinone pigments are preferably supplied for the pigmentation of plastics and high grade coatings.
1.3.3 Miscellaneous Pigments (Chapter 4)
There are a few pigment classes that can neither be sorted under hydrazone nor under polycyclic pigments. This group includes triarylcarbonium pigments and metal complex pigments as well as a number of individual pigments not belonging to a larger class of commercial pigments.
1.3.3.1 Triarylcarbonium Pigments (Section 4.1)
There are two groups of triarylcarbonium pigments: (i) inner salts of triphenylmethane sulfonic acids and (ii) complex salts with heteropolyacids containing phosphorus, tungsten, molybdenum, silicon or iron.
The first group is characterized by poor lightfastness and limited solvent resistance. Alkali Blue is the only member of this group with considerable commercial value. To tone black printing inks, Alkali Blue is used in combination with the very high-absorbing carbon black pigment, which increases its lightfastness considerably.
The second group includes the complex salts of basic pigments that are common in the dyes industry, such as Malachite Green, Methylene Violet, Crystal Violet or Victoria Blue with certain heteropolyacids. Despite the disadvantages of comparatively poor solvent resistance and limited lightfastness, these pigments are used for their excellent colour brilliance and clarity of hue – properties that exceed those of any of the other known organic or inorganic pigments. Such features make these types, whose lightfastness satisfies commercial requirements, suitable candidates for the printing inks industry and especially for packaging inks.
1.3.3.2 Metal Complex Pigments (Section 4.2)
Most metal complex pigments contain either an azo group (NN) or an azomethine (CHN) moiety. The metal is usually nickel or copper, and less commonly cobalt or iron(II).
Only a few azo metal complexes are available as pigments. They exhibit green or greenish yellow shades. Most of these are very lightfast and weatherfast.
Commercial azomethine complex pigments afford yellow, orange or red shades. Those species that provide the required lightfastness and weather resistance are used in automotive finishes and other industrial coatings.
1.4 Relationship between Chemical Structure and Pigment Properties
In this chapter, the correlation between chemical constitution and pigment performance is outlined in terms of empirical rules. These correlations are essentially applicable, independently of the application medium, to all industrial uses of pigments.
While the properties of (soluble) dyes are determined almost exclusively by their chemical constitution, application characteristics of pigments – which are by definition insoluble in the medium in which they are applied (Section 1.1) – are largely controlled by their crystalline constitution, that is, by their physical characteristics. This is discussed in the next chapter.
The application properties of a pigment are basically governed by its chemical constitution, which in turn has a bearing on the crystal structure, thus determining the physical parameters. This seemingly straightforward correlation is complicated by the fact that various crystal structures (modifications, see Section 1.5.3) may evolve from one and the same chemical constitution. Apart from knowledge about the chemical constitution of a compound, only extensive insight into the crystal structures and their solid-state physics thus allows certain predictions as to the application properties of the pigment.
This chapter discusses the influence of the chemical constitution on the hue, tinctorial strength, lightfastness, weatherfastness, solvent resistance, and migration resistance of a pigment. The systematic synthesis of a pigment with certain defined target properties is only possible to a very limited extent. Only with a reliable crystal-structure prediction can a prediction of the pigment's properties be made.
1.4.1 Hue
The appearance of colour in a molecule is associated with electronic excitation [5–9] caused by absorption of incident electromagnetic radiation in the ultraviolet and visible regions of the spectrum. Electrons are elevated from the ground state energy level to an excited state by absorbing selected frequencies of incident visible light, thereby giving the molecule the shade of the resulting complementary colour. The fact that each electronic excitation is accompanied by a battery of rotational and vibrational transitions is responsible for the appearance of more or less broad absorption bands. An absorption band is said to undergo a bathochromic shift if a comparison of spectra shows that it has moved to longer wavelengths; a hypsochromic shift involves movement to shorter wavelengths.
The hue is primarily defined by the pattern of chromophores, a conjugated π-system, which is responsible for the absorption of visible light. For transition-metal containing compounds, for example copper phthalocyanine, the electrons of the metal play a role, too.
Substituents with lone electron pairs, such as alkoxy, hydroxy, alkylamino and arylamino groups, are known as electron donors. Alkyl groups, despite the absence of such free electron pairs, are also considered to be electron donors. Functional groups with conjugated π-electron systems, such as NO2, COOH, COOR, SO2NH2 or SO2Ar, act as electron acceptors.
In discussing substituents of hydrazone pigments, both electron donors and electron acceptors are effective particularly as parts of the diazo component; that is, they are located in the conjugated part of the system. In these positions they usually cause a bathochromic shift of the absorption band with the longest wavelength. Evaluation of the empirical bathochromic effects of differently substituted derivatives against a standard conjugated system can provide a sequence of increasingly effective substituents. In azobenzene systems, for instance, electron donors are more bathochromically active than electron acceptors. Since this effect, however, also depends on the electron distribution between donor, acceptor and hydrazone function, a (usually less pronounced) hypsochromic shift may be observed, especially if electron acceptors are involved. Bathochromic or hypsochromic behaviour is also determined by the electron distribution at the substituted site within the conjugated system. If a bonding electron pair is promoted to a nonbonding excited state, a π → π* transition ensues that determines the frequency of the dominant absorption with the longest wavelength.
The intensity of an n → π* transition (transition of one electron of a lone electron pair from a nonbonding (n) to an antibonding (π*) orbital) is about two magnitudes less than the dominant π → π* transitions and thus has no effect on the colour.
Today, the Witt substitution rules, originally derived from empirical data on conjugated systems such as hydrazone derivatives, can be approached quantum mechanically. Hydrazone pigments basically obey the rules for azo dyes, although deviations through interactions within the crystal lattice must be taken into account.
The basic hue of a hydrazone pigment is primarily defined by the structure of the coupling component, since pigment manufacturers focus almost exclusively on substituted anilines as diazo components. Shades of yellow, for instance, are preferably produced by using acetoacetic arylides (CH3COCH2CONH-Ar) or heterocyclic coupling components based on the structure:
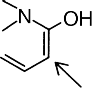
in a cyclic conformation. Products include barbituric acid (1) or 2,4-dihydroxyquinoline (2). Such compounds absorb mainly in the short wave (blue) region of visible light. The reddish yellow to orange shades produced by monohydrazone pigments obtained from 1-arylpyrazolone-5 derivatives (3) as coupling components have to be considered as exceptional.
A more bathochromic redshift is provided by monohydrazone pigments featuring the enlarged conjugated system of 2-hydroxynaphthalene (β-naphthol), particularly its 3-carboxylic acid and 3-carboxylic arylide derivatives.
Diazo components may also be of some significance in defining absorption frequencies. An amine with a considerably enlarged conjugated system, made, for instance, by dimerization, can contribute to a considerable bathochromic shift. Examples include the following:
| Diazo component | Coupling component | Shade | |
| Aniline derivatives | > | Pyrazolone-5-derivatives | Yellow to orange |
| 3,3′-Dichlorobenzidine | > | Pyrazolone-5-derivatives | Yellowish red |
| Aniline derivatives | > | Naphthol AS derivative | Red |
| 3,3′-Dichlorobenzidine | > | Naphthol AS derivative | Violet to blue |
Hydrazone pigments range in shade from greenish yellow to orange, red, blue, violet and brown. The chemical constitution of the pigment, especially the substitution pattern of the coupling component, determines the basic colour of a pigment; different shades within this colour are influenced by the crystal structure and by physical characteristics, such as particle size and morphology.
Substitution patterns, especially that of the diazotized aromatic amine, determine the colour of a pigment to some extent; however, empirical data do not lead to unambiguous conclusions as to the exact influence of a particular substituent on the shade. The problem is intricate, since the substitution pattern also has a bearing on the crystal structure, including all the interactions associated with it.
An exchange of substituents on the arylide moiety of the coupling component (arylides of acetoacetic acid and 2-hydroxy-3-naphthoic acid) fails to afford a consistent influence on the shade. In this case, intramolecular and intermolecular interactions within the crystal lattice gain more significance, since the conjugation of π-electrons is not expected to extend far beyond the carbonamide bridge.
The same general considerations apply to polycyclic pigments.
The insertion of heteroatoms into a polycyclic system, on the other hand, is frequently accompanied by a hypsochromic shift, due to a larger distance between the level of the highest occupied (HOMO) and the level of the lowest unoccupied molecular orbital (LUMO) (more exactly: due to the higher energy difference between the ground state and the ππ* state). Thus, for example:

The colouristic effect of substituting a polycyclic pigment system is expected to parallel that afforded by employing substituted diazo components in the manufacture of hydrazone pigments. Considerable chlorine substitution may even change the basic colour of a pigment. In contrast to the blue copper phthalocyanine pigments, copper polychloro-phthalocyanine pigments appear in green shades (Section 3.1); the chlorine atoms of tetrachloroisoindolinone pigments shift the absorption bands from the yellow region towards orange or red (Section 3.10).
The hue of a red hydrazone pigment lake carrying sulfonic acid functions is determined to a considerable extent by the metal ion. In the series of Na, Ba, Sr, Ca and Mn the shift of hue from yellowish to bluish red increases in the order in which they are listed. In addition, the polymorphic form and the hydration state may have a strong effect on the hue.
Particularly large colour changes are associated with the transition from solution to solid state in heterocycles such as the cross-conjugated indigo system or the quinacridone skeleton. Quinacridone pigments, dissolved in hot DMSO (dimethyl sulfoxide) or in DMF, exhibit a pale yellow shade; the intense red colour appears only in the solid state. This is a particularly distinctive example that demonstrates the correlation between crystal-lattice interactions and hue [10]. (For details see Section 1.5.3.)
Various methods have been developed to calculate and predict absorption spectra of hydrazone and polycylic compounds by quantum mechanics. However, the investigation of solid state spectra is complicated by interactions within the crystal lattice, which make accurate calculations of optical properties a challenging task.
1.4.2 Tinctorial Strength
Since the colour strength of a pigment is defined by its tendency to absorb light, the maximum molar extinction coefficient ɛmax can be used to estimate the relative strength of a pigment, which is, however, also dependent on the physical parameters of the pigment crystal (Section 1.5). A better approach is to measure the absorption connected with a single electron transition, especially the one with the longest wavelength, and to integrate the peak between the lower limit ν1 and the upper limit ν2 to calculate the total area f under the peak:
This is known as the oscillator strength. For practical purposes, ɛmax·Δν½, with Δν½ being the full-width at half-maximum, provides a good approximation. To be precise, the term oscillator strength refers to a free molecule or, as a first approximation, to a molecule in a solvent that does not noticeably interact with the chromophore system. Thus, for all practical purposes, it seems much more useful to approach the problem by focusing on the correlation between extinction and molecular parameters.
The primary chemical aspect of a mesomeric pigment system concerns the correlation between tinctorial strength and extent of electron delocalization. A higher degree of conjugation in a molecule is associated with a bathochromic shift; colour strength improves with the intensity of absorption. A resonating system proliferates by:
- enhancing the aromaticity of a polycyclic pigment, transition from monohydrazone to dihydrazone compounds (doubling of the hydrazone moiety in hydrazone pigments);
- promoting planarity in a molecule;
- incorporating substituents: π-electron interactions between the aromatic ring and adjacent conjugated free electron pairs, either nonbonding electron pairs or double bonds of substituents, improve electron delocalization, which in turn increases absorption intensity, that is, colour strength; the reverse is true for large substituents within a molecule that do not contribute to the resonating system – these may have an adverse effect on the tinctorial strength.
Crystal structure determinations point to a largely planar molecular structure in most pigments; this ensures optimum conjugation (overlapping of the π-electrons). Intramolecular hydrogen bonds contribute considerably towards supporting the planar conformation.
Molecular dimerization, as in the transition from Hansa Yellow to dihydrazone yellow pigments, increases the colour strength considerably. A comparison of a Naphthol AS/dihydrazone condensation pigment pair produces similar results:

G. Eulitz studied the correlation between chemical constitution and tinctorial strength directly on dissolved pigment samples [11]. He was able to exclude physical crystal parameters, such as particle size distribution and degree of dispersion, by achieving molecular dispersion. The transmission spectra of constitutionally similar pigment pairs, such as Pigment Yellow 1/Pigment Yellow 3, or the pair Pigment Yellow 12/Pigment Yellow 13, were evaluated according to the Lambert–Beer law.

The resulting extinction coefficient remained constant within the accuracy of the measurements. In the transition from monohydrazone to dihydrazone pigment, however, the value of the maximum molar extinction coefficient is more than doubled, because the effect of the two hydrazone linkages is enhanced by additional interaction via the biphenyl moiety. This, however, does not improve the energy difference between ground state and π π* state; the shade does not shift remarkably.
| Pigment solution | Maximum molar extinction coefficient (104 L/mol.cm) |
| Pigment Yellow 1 | 1.97 |
| Pigment Yellow 3 | 2.08 |
| Pigment Yellow 12 | 6.57 |
| Pigment Yellow 13 | 6.70 |
Quantum mechanical calculations showed that the extinction coefficients of these diaryl pigments strongly depend on the molecular conformation. If the biphenyl moiety is planar, ɛmax is much higher than in a twisted conformation [12].
Considering the fact that pigments exist as crystals dispersed in the application media, such studies on isolated molecules are generally of limited value. However, it has been shown that the maximum specific extinction of Pigment Red 112 is independent of whether it is measured in solution or in crystalline form. The study compared transmission spectra of the dissolved pigment with the solid-state spectra of crystals in a transparent film.
A certain correlation between the maximum extinction in solution and in the solid state has also been established for other classes of pigments.
1.4.3 Lightfastness and Weatherfastness
Little is known about the relation between chemical constitution and the lightfastness as well as the weatherfastness of pigments, although it is clear that fastness is primarily defined by the chemistry of the molecule. Various pigments, for example, undergo similar degradation upon exposure to light, irrespective of whether they are crystalline or in solution. The surrounding medium – the vehicle in which a pigment is applied – also affects the stability of a compound to light: the tendency of optically excited pigment molecules to react with their surroundings varies with the vehicle in which they are applied.
High-grade pigments cannot be studied both in solution and in the solid state, because they are insoluble in almost all solvents.
In hydrazone and some polycyclic pigments, a correlation may be established between the lightfastness and weatherfastness on the one hand and the substitution pattern on the other hand. Resistance to light and weather is generally improved by electron acceptors that are introduced through the diazo component (e.g. halogen atoms, nitro groups, carbo-alkoxy groups) – even more so in combination with electron donors (methoxy or methyl groups) in the phenyl moiety of the coupling component. Obviously, the substitution pattern of the aromatic system, especially that of the diazo component, has a major impact on the molecule: properties tend to improve in the sequence meta → para → ortho substitution. Ortho-substituents frequently take part in intramolecular hydrogen bonds.
The lightfastness and weatherfastness of quinacridone pigments (Section 3.2) deteriorate in the order 2,9 → 3,10 → 4,11 substitution. Decreasing the distance between substituents and NH function disturbs the formation of hydrogen bonds [13]; a tendency that culminates in the very poor light and weather resistance of N,N′-dimethyl-quinacridone.
The introduction of additional carbonamide groups frequently improves the lightfastness of hydrazone pigments, a trend that should not be confused with the generally observed tendency of carbonamide groups to enhance the migration fastness of their basic structure (Section 1.6.3). Monohydrazone yellow pigments, for instance, although more prone to migrate, survive exposure to light better than the almost insoluble diarylide yellow pigments. Dihydrazone condensation pigments, on the other hand, are much more lightfast and show less of a tendency to migrate than monohydrazone pigments.
Metal complex formation considerably improves both lightfastness and weatherfastness in o,o′-dihydroxyazo and o,o′-dihydroxy azomethine pigments, usually accompanied by a distinctly dull colour.
Likewise, the lightfastness of a hydrazone pigment lake is controlled to a certain extent by the metal cation: manganese salts are usually most lightfast.
1.4.4 Solvent and Migration Fastness
The influence of the chemical constitution as related to solvent and migration resistance opens up several options to improve pigment performance:
- Increasing the molecular weight
A comparison between related monohydrazone yellow pigments and diarylide yellow pigments clearly demonstrates the impact of an increased molecular weight on solvent and migration resistance. Pigment Yellow 12, for instance, migrates much less than a comparable monohydrazone yellow pigment such as Pigment Yellow 1. The fact that Naphthol AS pigments migrate much more readily than comparable dihydrazone condensation products adds to the evidence for the advantage of ‘doubling’ the molecular structure.
- Avoiding solubilizing substituents and adding insolubilizing moieties
Substituents such as long-chain alkyl, alkoxy or alkylamino groups as well as sulfonic acid functions tend to increase solubility.
Insolubilizing substituents are carbonamide groups and possibly nitro groups or chlorine for hydrazone pigments, hetero atoms, especially nitrogen and to a lesser extent also chlorine or bromine in polycyclic pigments.
The correlation between constitution (substitution pattern of the diazo component) and solubility of a hydrazone compound has been studied on a selection of Naphthol AS pigments [14]. With identical substituents, their position on the aromatic ring of the diazo component ultimately controls pigment solubility; that is, the difference in enthalpy between crystalline and dissolved pigment for a given substituent is largest for meta substitution and smallest for para substitution.
Polar substituents generally decrease the solubility of a pigment in common nonpolar solvents. Naphthol AS pigments, for instance, tolerate solvents much better and tend to migrate less easily than β-naphthol pigments (demonstrated by the pair Pigment Red 13/Pigment Red 3). The introduction of a second carbonamide function adds to the advantage, as in Pigment Red 170; this principle culminates in the production of almost completely migration resistant compounds such as Pigment Red 187. Solvent and migration fastness thus improve in the order shown below.

Consequently, there is a remarkable advantage in introducing carbonamide groups into various positions on heterocyclic five- and six-membered rings anellated to a benzene nucleus in order to improve the migration fastness of the basic structure (see structures 5–9). Of the various heterocyclic systems, the benzimidazolone moiety (5, X = NH), especially as part of the coupling component in an hydrazone pigment, has proven to be most effective, giving rise to an entire product line of benzimidazolone pigments (Section 2.8). Tetrahydroquinoxalinedione is used to a lesser extent (Section 2.8.5).

The supportive function of hydrogen bonds towards decreasing the solubility of a pigment is particularly striking in benzimidazolone pigments: the good solvent and migration resistance of these pigments compared to their monohydrazone yellow and Naphthol AS counterparts [15, 16], see also Section 2.9) relies to a large extent on the comparatively small benzimidazolone group and its capacity to form intermolecular hydrogen bonds. The same is valid for tetrahydroquinoxalinedione pigments (Section 2.8.5).
A combination of these first two options (a and b) is possible as well. For example, the combination of ‘doubling’ the molecular structure and introducing benzimidazolone groups leads to the extremely insoluble P.O.72 (Section 2.8.4).
- The formation of insoluble polar salts by laking
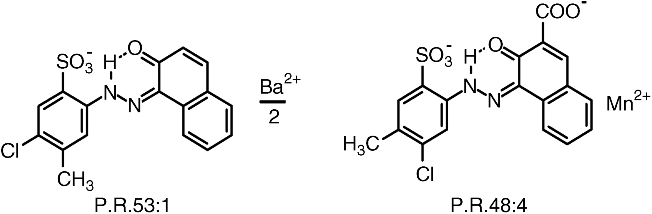
Azo dyes containing sulfo and/or carboxy groups form pigment lakes if they are precipitated with the salts of alkaline-earth metals such as calcium, barium, strontium or with manganese. The polar (salt) character of such pigment lakes makes for good solvent and migration fastness. Consequently, an increasing number of sulfo groups for salt formation is associated with improved solvent fastness, as demonstrated by Pigment Red 53:1 and Pigment Red 48:4:
- The formation of metal complexes
Complexation of o,o′-dihydroxyazo compounds or their corresponding azomethine derivatives to metals affords extremely solvent and migration resistant pigments (Section 4.2).
1.5 Physical Characterization of Pigments
The relevance of the chemical constitution of a pigment in controlling its application properties (Section 1.4) is embedded in the context of physical parameters, such as geometry of the unit cell, crystal lattice pattern (modification) and crystal shape, along with particle characteristics such as specific surface area, crystallinity, particle size distribution and surface structure. Pigment properties as defined by certain target applications can be approached or optimized by adjusting the physical parameters. Particle size modifications, for instance, frequently facilitate the use of a pigment in an area in which its applicability would normally be compromised by processing problems or in which the pigmented medium might exhibit poor application properties.
A significant comparison of the application properties of pigments with different chemical constitutions can only be meaningful if it focuses on compounds whose physical characteristics are somewhat similar (particularly particle size).
There is an increasing tendency amongst manufacturers to provide their pigments with physical specifications. This facilitates comparisons between pigments and provides the user with some indication about application properties that may be relevant for their specific needs. Considering the importance of such information, it seems useful to discuss a few physical characteristics and methods for their determination, including the inherent limitations of such methods. The discussion focuses on organic pigments and the implications of the resulting data to the pigment chemist. There are several publications and text books that can familiarize the reader with the underlying theory and the application of such methods [17].
All pigments have some degree of particle size distribution. The ultimate particle size of most commercial pigments is less than 1 µm, often even smaller than 0.3–0.5 µm (anisometric particles are measured lengthwise). Depending on the application for which a pigment is targeted, the smallest particles may be as much as one to more than two orders of magnitude smaller.
For surface energy reasons, the tendency of these small particles to agglomerate and form crystallites increases with decreasing particle size. This is particularly true for the final phase of pigment manufacture, the drying and milling processes.
Organic pigment powders therefore consist of a mixture of such crystallites and single crystals. Incorporation into the application medium, be it a plastic, a printing ink or a paint, relies on dispersion, which involves an effort to break down the agglomerates as far as possible. The quality of the pigment dispersion determines the majority of the application properties of a pigment, especially regarding the tinctorial and rheological potential of a pigment–vehicle system (Section 1.6.5).
The particle size distribution of an organic pigment powder is usually different from that found in the pigment–vehicle system; and since both have practical importance, methods have been developed for their determination.
Pigments are morphologically described by a standardized terminology [18]; they can occur as single or primary particles, aggregates or agglomerates.
The final crystals that ultimately constitute the crude pigment product are known as primary particles. These are true single crystals with the typical lattice disorders or combinations of several lattice structures that appear as units under X-ray analysis.
Primary particles may assume various shapes, such as cubes, platelets, needles or bars, as well as several irregular shapes. Figure 1.1 shows some examples. Combinations between different shapes often make the unequivocal assignment to a particular class impossible.

Figure 1.1 Examples of crystal shapes of organic pigment particles.
Aggregates are primary particles that are grown together at their surfaces. The total surface area of an aggregate is smaller than the sum of the surfaces of the individual particles. Aggregates cannot broken down by dispersion processes.
Agglomerates are groups of single crystals and/or aggregates, joined at their corners and edges but not grown together (Figure 1.2), which can be separated by a dispersion process. The surfaces of the individual crystals are readily available to adsorption. By definition, the total surface area of an agglomerate may not differ considerably from the sum of the surfaces of the individual particles. The dispersibility of a pigment is largely determined by the nature and density of the agglomerates, which in turn depend on particle shape and density. Platelets or rodlike particles, for instance, generally make for more voluminous and more easily dispersible agglomerates than isometric species.

Figure 1.2 Primary particles, aggregates, and agglomerates according to DIN 53206, part I [18].
Pigment particles that are already dispersed for various reasons may reassemble and form loosely combined units with various shapes. The most important among these are flocculates (Figure 1.3) – assemblies of wetted crystallites and/or aggregates or smaller agglomerates. They usually form in a low viscosity medium which fills the interior cavities of the pigment flocculates. Flocculates are therefore mechanically more labile than agglomerates and can usually be broken up by weak shear such as stirring.

Figure 1.3 Pigment Red 3 in an air-dried alkyd resin film. (a) Electron micrograph of an ultramicrotome-cut thin layer of a pigment flocculate; (b) magnified image.
1.5.1 Specific Surface Area
The specific surface area of a pigment is the surface in m2 per gram of pigment. Typical values for organic pigments range between about 10 and 130 m2 g−1.
The surface area of a pigment, however, is not a definitive value: it is controlled by factors such as the method of determination and the experimental parameters. Depending on the procedure, the experimental results may either reflect the exterior geometry of the pigment particles or also account for additional interior surface area, which is hard to define. Parameters such as the molecular size of the adsorbed substance and pigment geometry, especially its porosity, determine how much of the total surface area is accessible to measurement.
Adsorption techniques are based on the assumption that the amount of gas, liquid or solute that is adsorbed by a surface is proportional to the surface area. The resulting linear correlation makes it possible to calculate the total surface area of a pigment from the molecular volume of the adsorbed material and the amount that is necessary to completely cover the surface with a monolayer. The amount of adsorbate may be determined:
- directly from the pigment sample,
- from the change of pressure in the adsorbed gas,
- from the concentration of the adsorbate dissolved in a carrier gas or in a liquid,
- from the heat of reaction produced or required by the adsorption process.
To determine the surface area of a pigment, it is more common to enforce an adsorption process by working at low temperatures and high pressure and concentration than to rely on desorption, which can be brought about by reversing the conditions.
The BET method, an adsorption method working with nitrogen, argon or krypton gas, is the most widely used technique to measure specific surface areas of organic pigments. The isotherms are evaluated according to the Brunauer, Emmett and Teller (BET) equation [19], which is decribed satisfactorily in text books [20–23]. The market of routine determinations is dominated by commercial instruments that focus on a single point on the adsorption line close to the upper limit of the BET equation instead of plotting the entire adsorption isotherm.
Likewise, the Haul and Dümbgen method of measuring specific surface areas through nitrogen adsorption has developed into an industrial standard [24].
The interior surface area of a pigment powder may be assessed through a series of determinations, using a range of gaseous, liquid or dissolved substances of various molecular sizes with different spatial demands, and measuring the amount of adsorbed material gravimetrically as well as volumetrically [25].
Several phenols have proven to be valuable adsorbates for surface area determinations in organic pigments [26]. Phthalocyanines in particular have been evaluated successfully by phenol adsorption [27].
All of the gas adsorption methods rely on the availability of the entire interior surface of the pigment, including the inside of the agglomerates, to the gas molecules. In organic pigments, however, this is not always true, so that the results often seem considerably smaller than the actual values. Two varieties of Pigment Red 168 whose surfaces had not undergone treatment prior to the experiment have been used to demonstrate this effect [28]. Superior hiding power in sample 2 seemed to be connected with a larger specific surface area. Initial measurements afforded values of 20.8 and 36 m2 g−1 for samples 1 and 2, respectively, which remained essentially constant even after the samples were rinsed thoroughly with organic solvents. Interestingly, the BET results contradict the electron microscopic photographs of the two samples (Figure 1.4), which indicate that the particle size in sample 1 is relatively small, whereas sample 2 appears to be comparatively coarse.

Figure 1.4 Electron micrographs of Pigment Red 168. The pigment was dispersed in ethanol by means of ultrasound and vaporized onto the electron microscope slide. Sample 1: 20.8 m2 g−1 (a); sample 2: 35.9 m2 g−1 (b).
The experimental results are in keeping with the tendency of the agglomerates of sample 1, which are unavailable to wetting by polyolefins, to resist dispersion in nonwetting application media even in the presence of vigorous shear. Sample 2, however, disperses easily under the same conditions, which considerably improves its tinctorial strength. The agglomerates in sample 1 can be broken down by predispersing the sample in a molten low molecular weight polyethylene wax and simultaneously applying shearing force to the material before incorporating the pigment into the polyolefin. Unsurprisingly, after the same treatment has been applied to both samples, the tinctorial strength and transparency of sample 1 exceeds that of sample 2. A similar effect occurs in the process of slurrying both samples in dioctyl phthalate and subsequently working them into plasticized PVC.
Clearly, there is little sense in experimentally determining specific surface areas of highly agglomerated organic pigment powders with comparatively small particle sizes; it is not advisable to rely on such values alone. Unfortunately, since both manufacturers and the literature fail to provide specifications about degrees of agglomeration in commercial products, the user faces the problem of dealing with values for specific surface areas that give little or no indication of the application properties of a particular product.
The same effect arises from a phenomenon known as ‘overmilling’ organic pigments: inadvertently reducing both specific surface area and tinctorial strength of a pigment powder by milling it for too long or too intensely, so that particles are fractured, only to reassemble and produce increasing amounts of agglomerates.
Agglomeration, however, is not the only factor to falsify the experimental results of surface area determinations; there may be several substances adhering to the surface of the pigment particles that affect gas adsorption. Hydrazone pigments, for instance, are manufactured using auxiliary agents, such as emulsifiers, coupling agents, and so on (Section 2.2), which are partially adsorbed on the fresh surface of a newly synthesized pigment crystal. Measurements may therefore reflect specific surface areas that appear smaller than the expected value.
Frequently, large amounts of additives such as rosin are used in manufacturing highly transparent types of hydrazone pigments for application in process colour printing inks. Pigment particles that emerge from such a process are comparatively small, because such resins tend to inhibit crystal growth [29]. Although they may occur separately, these substances are largely adsorbed on the surface of the pigment particles, where they interfere to a greater or lesser extent with the surface area determinations.
The extent to which measurement results can be distorted by additives is particularly severe in the case of dishydrazone yellow pigments, whose preparation involves fatty amines (Sections 1.8.1.1 and 2.4.1.1). Measurements are commonly carried out under conditions that give rise to the cleavage of volatile amines or ammonia, even resulting in seemingly negative specific surface area values.
The extent to which surface area determinations are distorted by traces of water that cling to the particles, despite extensive drying, is unknown.
There are also techniques that utilize flow phenomena to measure parameters such as surface area or average particle size. Information about the latter is obtained by analyzing the amount of gas (air) or wetting liquid that is pulled or pressed through a defined pressing of pigment powder. This approach is based on the principle that the flow rate is controlled by the interior cavities of the pressing, which in turn depend on the particle size: the larger the particles, the higher the number of sizable cavities. The experimental results obey the Carman–Kozeny equation [30], which may be used to calculate the specific surface area. Surface-to-volume ratios of less than 1.2 m2 cm−3 may be determined by permeability techniques, amongst which the Blaine method and instrumentation has developed into an industrial standard in Germany [31]. Flow methods tend to be unsatisfactory, however, in connection with organic pigments. Extremely large specific surface areas and exceptionally wide particle size distributions compared to inorganic pigments and extenders provide ample opportunity for surface coating materials and other surface processing additives to distort the measurements.
It seems reasonable to conclude that experimentally determined specific surface areas can only qualitatively relate to the physical characteristics of organic pigments. Instead, their value emerges in combination with other physical or physico-chemical parameters or in the context of application properties such as oil absorption [32] or wettability (Section 1.6.5).
1.5.2 Particle Size Distribution
The particles of a synthetic pigment, far from being uniform, cover a more or less wide range of sizes. Normally discontinuously produced pigment batches are usually combined so as to yield mixtures that meet the technical standards of certain target applications. This explains why it is possible for different batches of the same pigment to exhibit somewhat divergent particle size distributions.
There are various methods for the determination of the size distribution of organic pigment particles, the most common being sedimentation techniques in ultracentrifuges and specialized disk centrifuges as well as electron microscopy. These methods require considerable experimental skill, since the results depend largely on sample preparation and especially on the quality of the dispersion.
The agglomerates that constitute organic pigment powders (Section 1.5) are more or less broken apart during the dispersion process, for example incorporation into the application medium, which leaves a pigmented system consisting of primary particles, aggregates and smaller agglomerates. The dispersion process is a very complex phenomenon and its outcome depends largely on various factors (Section 1.6.5). It is therefore not surprising that the degree of pigment dispersion that is achieved under certain conditions is by no means constant. Experimentally determined particle size distribution information can only relate to the application properties of a pigmented system if the particle size distribution of the pigment powder exhibits roughly the same values as in the pigment–vehicle system. This, however, unfortunately poses something of a challenge.
Size analyses are commonly carried out by mixing the pigment powder with an organic solvent or with water and adding appropriate surfactants to enhance the dispersibility of the powder. Aqueous dispersions frequently undergo size separation in ultracentrifuges, while organic solvents are more appropriate for electron microscopic techniques.
1.5.2.1 Determination of Particle Size by Ultrasedimentation
Discrete particle size separation or fractionation is carried out in the strong gravitational field of rapidly rotating ultracentrifuges. The theoretical background and experimental technique are described in depth in the literature [33–35]. While there is some difficulty in preparing a completely homogeneous pigment suspension, the two-layer version of the Marshal method suffers particularly from separation problems, including a phenomenon known as the streaming effect*.
The different ball mills that are currently used to prepare aqueous pigment suspension for sedimentation analyses share the disadvantage that mutual abrasion between particle, ball, and container surface can hardly be avoided. Suitable corrections must also be applied to account for incomplete and inconsistent dispersion of the very fine agglomerates of organic pigment particles through ultrasonic dispersion techniques. Ultrasound dispersion is therefore often associated with a certain trend towards larger portions of coarse particles at the expense of the finer grains. As a result, the pigment powder that is dispersed by the ultrasound technique is not dispersed to the same degree as in its application medium. The values for comparatively coarse pigments that disperse easily in aqueous media agree much better. No such problems exist for specialized pigment preparations in which the pigment is predispersed so as to distribute easily in the medium of application without extensive shear, such as emulsion paints. With these, even post-preparative flocculation can safely be neglected [36]. The degree of pigment dispersion in the analysed suspension is thus the same as in the preparation and therefore in the medium of application.
Particle size distribution analyses in organic solvents may show dissolution and recrystallization effects when pigments with poor solvent fastness are measured.
There are several methods by which to monitor the sedimentation process, but optical techniques have prevailed since the 1970s. Joyce Loebl [37] paved the way for this analytic method by designing transparent disk centrifuges. Today it is possible to study organic pigments by photosedimentometry with conventional ultracentrifuges. The entire particle size distribution is therefore determined during one sedimentation process only, while older methods required a separate gravimetric analysis for each point on the distribution curve.
The pigment concentration in a photosedimentometry sample is very low. The pigment suspension is placed into a small cell that is inserted into a rotor. By directing a beam of light through the windows of the cell, a transmission–time curve is recorded during the run of the centrifuge, which provides particle size distribution information through the quantitative correlation between specific extinction coefficient and particle size. White light is most commonly used, although it may be replaced by a monochromatic source or a laser beam [38]. Photometric methods are not susceptible to reagglomeration, because the experiments are carried out at pigment concentrations as small as 10−5 % by weight.
Recent ultrasedimentation techniques for pigment analysis have abandoned the two-layer principle. Measurements can now be taken directly on the aqueous dispersion itself, which alleviates several problems, such as the streaming effect. However, both the intricate instrumentation and the complex mathematical evaluation require considerable skills as well as familiarity with sedimentation theory and practice.
1.5.2.2 Determination by Electron Microscopy
The difficulties that accompany the dispersion of a pigment for ultrasedimentation analysis (Section 1.5.2.1) parallel those associated with electron microscopy with its high demands on sample preparation.
Even a routine operation, such as applying the pigment suspension to a microscope slide by ultrasound vaporization, may be a source of complications. Pigment particles, previously completely dispersed in suspension and even in a vaporized droplet, may reagglomerate in the suspension whose volume is reduced as the carrier (organic solvent or water) evaporates.
There are several organic solvents whose value as dispersants, although they may be more suitable than water or alcohol, is compromised by side effects. Such agents may dissolve materials such as collodion, which is used as a thin film on support grids, or they may distort the particle size distribution by promoting recrystallization during dispersion and application. Such solvents are therefore not useful for the electron microscopic analysis of organic pigments.
It seems more important to focus on problems that may arise during quantitative evaluation of electron photomicrographs of an organic pigment. Automatic image analysers cannot extract information from images that indicate more or less agglomerated, nonisometric or even platelet-shaped particles.
Particles arranged on top of each other can be detected individually by semi-automatic analysers; but the difficulty remains with studies of agglomerated units, and results tend to be inaccurate.
It is obviously not easy to measure the variable dimensions of platelets, rodshaped or acicular particles that differ in length, width and height. The shape outlined in the image reflects the orientation of a particle relative to the analyzing beam; a graphical grid allows two-dimensional examination of such a profile. The third dimension, however, which illustrates particle size and shape, remains elusive. Several approaches to a more complete image interpretation introduce additional factors to represent the particle shape; however, they are accompanied by a fresh set of problems. One technique [39, 40] affords comprehensive two-dimensional or three-dimensional descriptions of anisometric particles through scattering ellipses and/or ellipsoides.
Scanning electron microscopy is a common and very useful instrument in determining particle size distributions in organic pigments; its advantage lies in the recognition of individual particles lying on top of each other as well as horizontally agglomerated units. Depending on whether the graphical grid is applied to a photograph obtained by TEM or by SEM techniques (Figure 1.5), it is therefore possible for a pigment sample to display different particle size distributions (Figure 1.6).

Figure 1.5 Images of samples of the same organic pigment, taken with a transmission electron microscope (a) and a scanning electron microscope (b) at equal magnification.

Figure 1.6 Particle size distributions of the same pigment sample as derived from images taken with a transmission electron microscope (a) and a scanning electron microscope (b).
Pigment particles to be studied by electron microscopy are sometimes incorporated into ultrathin membranes. At first glance, quantitative image analysis of such ultrathin layers seems convenient, since it appears to reflect the distribution of pigment particles in the medium in which they are applied. This argument is as compelling as it is deceptive, because during the preparation of the ultrathin layers both isometric and platelet-shaped or acicular crystallites are severed at random and ultimately reproduced in various spatial orientations (Figure 1.7).

Figure 1.7 Cross-sections through rod-like pigment particles in relation to the cutting angle.
Depending on the method of application, platelets or other pigment particles may be orientated along a certain axis when they are dispersed in their medium. In this case, the values obtained by electron microscopy are largely determined by the geometric arrangement of the acicular pigment particles within the sample membrane; that is, the results depend on whether their orientation is parallel or perpendicular to the plane of the ultrathin layer (Figures 1.8–1.10).

Figure 1.8 Electron micrograph of an ultramicrotome-cut thin section of a layer of paint, seen parallel to the orientation of acicular pigment particles and perpendicular to the surface of the layer.

Figure 1.9 Electron micrograph of an ultramicrotome-cut thin section of a layer of paint, seen perpendicular to the orientation of acicular particles and to the surface of the layer.

Figure 1.10 Electron micrograph of an ultramicrotome-cut thin section of a layer of paint, seen parallel to the orientation of acicular pigment particles and to the surface of the layer.
A phenomenon known as the rub-out effect (Section 1.6.5) frequently evolves from a certain set of conditions. Most often, it occurs during the application of a paint film. Particle size separation occurs when pigment particles accumulate along the substrate or on the surface of the coating, reversing the dispersion process. A suitable experimental technique must account for this effect by considering the entire layer.
There are various problems that might be solved if the particle size distribution within the medium of application can be elucidated. This is especially true where the undiluted paint system is used. In 1968, Geymeyer and Grasenick of the Technische Hochschule in Graz broke new ground in this area by proposing a new experimental procedure, a novelty at the time. They designed specialized equipment to rapidly cool the liquid pigment–vehicle system to very low temperatures by inserting it into liquid nitrogen, where the material is dissected into ultrathin layers. The resulting sections are freeze-dried to remove all traces of solvent and may then undergo etching with nascent oxygen. Finally, vaporization techniques are used to cover the dried sections with thin layers of graphite and gold for observation with a scanning electron microscope. During the initial phase of the cooling process, however, interaction between pigment particles or between pigment and vehicle particles tends to result in a certain degree of aggregation. The structure of these units is a function of the pigment and the vehicle composition (Figures 1.11–1.13). Methods to reliably predict the exact distribution of a pigment in its carrier thus remain to be developed.

Figure 1.11 Pigment Yellow 3 in an airdried alkyd resin paint.

Figure 1.12 Pigment Red 53:1in a publication gravure printing ink based on calcium resinate.

Figure 1.13 Pigment Yellow 12 in a publication gravure printing ink based on metal resinate.
Particular difficulties similar to those associated with specific surface area determinations (Section 1.5.1) also accompany particle size distribution studies on surface coated organic pigments.
The identity of the additive, be it an amine, a hard resin or some other material, is less of a concern than the question of its concentration. There is no information on the concentration limits above which an additive may distort the measurements, but one can expect this value to be defined largely by the specific surface area and the average particle size of each individual pigment. For pretreated surfaces, sizing the pigment particles by electron microscopy thus appears more reliable than determining the particle size distribution by ultrasedimentation. The additive typically functions as an adhesive that attaches pigment particles to each other, releasing them by dissolution processes only through dispersion, which more or less distributes the pigment throughout the application medium. It is therefore impossible even for powerful dispersing agents to break down such units in an aqueous suspension for ultracentrifugation purposes. Electron microscopy, on the contrary, allows the use of resin-dissolving agents to facilitate dispersion. The only disadvantage is that artefacts may develop during evaporation.
Other instrumentation used to determine particle size distributions, such as the coulter counter, tends to be unreliable and problematic for organic pigments. This is also true for methods such as analysing laser light passing through a pigment dispersion. The intensity of the resulting light pulses reflects the frequency of Brownian motion within the sample, which in turn is a function of the particle size. This technique works for particle sizes between 40 and 3000 nm.
1.5.2.3 Data Representation
Particle size distribution may be characterized in terms of a spread of values in a table, a graphical representation in the form of a histogram (block diagram) or as a continuous curve (Figure 1.14) [41].

Figure 1.14 Representation forms of particle size distributions.
Nonlinear representations allow plotting of the data such that particle size distributions are well arranged and facilitate interpolation and extrapolation. The most common amongst these are defined by standards, such as the linear and the logarithmic representation for distribution sums [42], the exponential system [43], the logarithmic normal distribution [44] and the RRSB grid, which derives its name from the authors Rosin, Rammler, Sperling and Bennett [45]. This list also includes:
1.5.2.3.1 Density Distribution
The percentage of particles with the same given parameter is plotted against this parameter. A property that lends itself excellently to this purpose is the equivalent spherical diameter (D), which is the diameter of a fictitious spherical particle. The parameter determined by sedimentation techniques is usually the equivalent spherical diameter or the Stokes diameter (Dae and DST, respectively), which is the diameter of a sphere of equal density that would have the same sedimentation velocity in the medium as the nonspherical particle being measured. Another commonly used property is the light scattering equivalent diameter, DL, which is the diameter of a sphere that has the same light scattering ability as the particle in question. All these parameters, however, should be treated with some caution, because the concept of the equivalent spherical diameter is based on an ideal spherical particle, which remains fictitious. According to one study [11], however, laminar flow effects that prevail during sedimentation render the velocity of rod-shaped particles in a medium independent of their orientation.
1.5.2.3.2 Sum Distribution
A cumulative presentation of equivalent diameters, which converts the density distribution curve into a plot that represents percentages of particles that are smaller than a given equivalent diameter D.
It is possible to graphically present both the density distribution and the sum distribution according to either one of the following mathematical representations (Figure 1.15):
1.5.2.3.3 Numerical Distribution
n(D), the percentage of particles with a given equivalent diameter.
1.5.2.3.4 Surface Area Distribution
s(D), the percentage of particles with a certain surface area plotted against the equivalent diameter.
1.5.2.3.5 Volume or Mass Distribution
v(D), the percentage of particles with a certain volume or mass, respectively, plotted against the equivalent diameter.

Figure 1.15 Numerical distribution, surface distribution and mass distribution.
The choice of method depends both on the purpose for which the representation of the particle size distribution is intended and on the experimental technique by which it is determined.
The numerical distribution is important for organic pigments; this is particularly true for electron microscopy, which affords results in terms of numbers of particles (Section 1.5.2.2).
Surface area distributions are especially adapted to reflect the dispersion characteristics or solvent resistance of pigment particles, since properties such as surface energy and dissolution rate are proportional to the surface area of a particle. Lightfastness and tinctorial features of pigment particles, such as tinctorial strength, hiding power or transparency, are best compared by using mass distribution or volume distribution representations. This is true because the extinction of an organic pigment powder with a particularly fine particle size is largely proportional to the mass of the particles. The different mathematical representations of particle sizes are interconvertible.
There is a standardized terminology to designate the mean values for the different particle size distribution representations [18]. Amongst these are (Figure 1.16):
- The arithmetic mean (Da). This is the mean diameter of either the numerical (Dan), the volume (Dav) or the surface area distribution (Das).
- The most frequent diameter Dmf (the mode). This is the diameter at the maximum of the frequency distribution.
- The median or central value Dc = D50%. This is a cumulative value that represents the diameter that lies exactly between the upper half and the lower half of all particle sizes and thus corresponds to a sum distribution of 0.5 = 50%.

Figure 1.16 Different mean values for the particle diameter D.
Despite the fact that all of these values are quoted freely in the literature, it is only the median that seems to be of any physical significance, considering the typically unsymmetric particle size distributions of organic pigments.
Standards have also been developed for the statistical evaluations of frequency or numerical distributions, means and standard deviations, confidence intervals, regression and correlation [41].
1.5.3 Crystal Structure and Polymorphism
The crystal structure has a major impact on the pigment's properties. In principle, all solid-state properties of organic pigments depend on the crystal structure. This includes:
- optical properties like colour, hue, tinctorial strength, cleanness and hiding power;
- crystal growth rate, crystal morphology, and crystal sizes; mechanical processability, rheology in dispersions;
- surface properties of the crystals; agglomeration and aggregation behaviour, flocculation, dispersibility;
- thermal stability, phase-transition points, melting point;
- density;
- crystallinity;
- solubility and rate of dissolution (which are also a function of the particle size), solvent and migration fastness and bioavailability;
- chemical stability;
- light and weather fastness (Section 1.7.4).
Most organic pigments can exist in different polymorphic forms, that is, crystal structures with different arrangements of molecules, resulting in impressive colouristic variations. For example, the α-phase of P.O.36 is brown, whereas the β-phase is orange; the α-phase of P.Y.213 is yellow, but the β-phase is brown; P.O.72 has orange, red and brown polymorphs; the individual polymorphs of P.V.19 and P.B.15 differ in their shades and their fastness properties.
Historically, polymorphism of organic colourants was observed as early as in 1933, when scientists at BASF found four different polymorphic forms of Sudan Orange R (coupling product of diazotized aniline with β-naphthol). The polymorphic forms, which have similar colouristic properties, were detected by X-ray powder diffraction [46]. In 1934 onwards the polymorphism of indanthrone (P.B.60) was investigated at BASF, and four polymorphs were found [47].
A profound understanding of a pigment's property requires knowledge of the crystal structures and of the structure–property relationships as well.
Organic pigments molecules are very efficiently packed in the solid state. The lattice energy is large, which explains the insolubility of the pigments. Calculations show that the lattice energy consists mainly of van der Waals interactions, supported by hydrogen bonds and Coulomb interactions between partially charged atoms. In laked pigments ionic interactions between the metal ion and the organic ligands play a major role.
Frequently, the planar pigment molecules are stacked, and form piles. However, the molecules never stack exactly on top of each other. They are always laterally shifted, so that the atoms of one molecule fall into the hollows of the van der Waals surface of the neighbouring molecule; thereby, the packing density and the strength of the van der Waals interactions are optimized. A special π–π interaction does not exist in any organic pigment; the frequently observed ‘π-stacking’ of pigment molecules is favoured by a high number of van der Waals (and Coulomb) interactions.
The efficient molecular packing results in high densities, up to 1.71 g cm−3 for the perylene P.R.224 (C24H8O6) [48], which is amongst the highest densities for all organic compounds consisting of C,H,N,O atoms only. The introduction of halogen or sulfur atoms leads to even higher densities, for example 1.88 g cm−3 for tetrachlorothioindigo, P.R.88 (C16H4Cl4O2S2) [49]. Hydrazone pigments tend to have lower densities than polycyclic compounds.
The crystal structures of the individual pigments are discussed in connection with the corresponding classes of pigments (Sections 2.3 to 4.5).
1.5.3.1 Effect of the Crystal Structure on the Optical Properties
The optical properties of organic pigments, especially colour, hue, tinctorial strength, purity of shade and hiding power, strongly depend on the crystal structure. (For an overview, see Reference [50].) The impact of the crystal structure on the optical properties can be separated into four main factors: hydrogen bonding, exciton coupling, molecular conformation and crystal morphology:
- Hydrogen bonding
Hydrogen bonding has an influence on the electronic structure of the pigment molecules, especially if the hydrogen bonds are formed with atoms of the chromophoric system. For example, individual molecules of quinacridone (P.V.19) have a yellow shade, which is caused by the weak conjugation between the three benzene rings (Figure 1.17a). This yellow colour is visible in a very dilute hot solution of P.V.19 in DMSO. In the crystalline state the molecules are connected by hydrogen bonds, which result in weakening of the NH and CO bonds, and an increased conjugation between the benzene rings (Figure 1.17b). Correspondingly the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is reduced; more exactly, the excitation energy for the π → π*-transition is reduced. For individual molecules, violet light is absorbed, resulting in a yellow colour. For hydrogen-bonded molecules, green light is absorbed, resulting in red shades [51]. The intermolecular hydrogen bond network depends on the crystal structure of the individual polymorph, which may result in different shades for the individual phases. For other pigments, the hydrogen bond topology within the molecule, too, can be affected by the molecular packing, as was found for P.Y.213 [52].
- Exciton coupling (interaction between transition dipole moments)
The excitation energy depends not only on the molecule itself but also on the relative positions and orientations of all neighbouring molecules, even of those that are not connected by π-stacking or hydrogen bonds (exciton coupling). The electronic excitation of a molecule can be described by a transition dipole moment. In a first approach the exciton coupling is described as a classical dipole–dipole interaction between the transition dipole moment of the molecule and the moments of neighbouring molecules.
The effect of exciton coupling on the colour is clearly visible in a series of perylene pigments:

The substituent R has only a minor effect on the π-system of the perylene, because the phenyl rings are almost perpendicular to the perylene moiety, thus prohibiting electronic conjugation with them. Nevertheless, the colour of these perylene pigments varies from scarlet-red (P.R.123) via bluish red (P.R.190) to black (P.Bl.32). The non-commercial pyridylethyl derivative has three crystal phases with red, reddish black and black colours. Obviously, the colour is determined not by the electronic effects of the substituent R, but by the crystal structure. The relative position of the perylene moieties of neighbouring molecules differs strongly in the series of perylene pigments. Based on the structures of 20 perylene compounds, an empirical correlation between the relative position and the colour was established in 1989 [53]. More recently, it has been shown that the differences in the arrangement of neighbouring molecules cause considerable differences in the exciton coupling, which explains the colour changes [54].
- Molecular conformation
The optical properties also depend on the conformation of the molecules in the solid state. The conformation can strongly be affected by the molecular packing in the lattice. For example, the colour strength of diaryl pigments depends on the twisting of the central biphenyl unit; increasing twisting leads to decreasing conjugation between the two π-systems and a reduced colour strength. P.Y.12 and P.Y.13, which differ only by four methyl groups on the terminal phenyl rings, have a similar colour strength in solution. In the solid state the twisting is controlled by the molecular packing: In the commercial α-phase of P.Y.12 the biphenyl unit is twisted by 27°, whereas the molecules of P.Y.13 are planar in the solid state. Correspondingly, the colour strength of the α-phase of P.Y.12 is significantly lower than that of P.Y.13. The difference in molar extinction coefficients is caused solely by the different crystal structure [55].
- Crystal morphology
Crystal size and morphology, which strongly depend on the crystal structure, have a major impact on the colouristic properties, too (Section 1.7).

Figure 1.17 Increasing conjugation in P.V.19 due to hydrogen bonding in the solid state.
Furthermore, the vibrational states also depend on the molecular conformation, the hydrogen bond network and the arrangement of neighbouring molecules. This affects the width and shape of the absorption bands, thereby modifying the optical properties as well.
1.5.3.2 Polymorphism
Polymorphs are solids that have an identical chemical composition but differ in the arrangement of molecules in the solid state, that is, in their crystal structures. The molecular conformation can be different, too. There are even cases in which the unit cell simultaneously contains two pigment molecules with different conformations [56].
Additionally, the crystal lattice can contain water or solvent molecules, leading to hydrates and solvates. Some pigments even form solvate-hydrates or mixed solvates. (For example, one of the non-commercial phases of P.Y.181 contains one DMSO and one NMP (N-methyl-2-pyrrolidone) molecule per pigment molecule in the unit cell [57].) At present there are no commercial pigments with solvent molecules in their crystal lattice.
Polymorphs are also called ‘modifications’. Polymorphs, hydrates and solvates are summarized as ‘crystal phases’. Strictly speaking, ‘polymorphs’ are crystal phases with exactly identical chemical composition, whereas crystal phases that differ in their water or solvent content are called ‘pseudopolymorphs’. Formally, hydrates with different amounts of water molecules in the crystal lattice should not be named ‘polymorphs’ but ‘pseudopolymorphs’. However, the number of water molecules in the crystal lattice is not always known, and in the pigment literature (as well as in the pharmaceutical literature) hydrates with different water contents are included in the term ‘polymorphs’. In this book we follow this usage.
Commercially used pigments generally do not contain water molecules in the crystal lattice. Laked pigments are an exception. They frequently form hydrates. Crystal phases of laked pigments may differ in their water content. For example, the α-phases of P.Y.183 and P.Y.191 are monohydrates while the corresponding β-phases are trihydrates; all four phases are commercially used. As in in several other laked pigments, the water molecules are bonded to the metal ion. This bond can be very strong, leading to a high thermal stability. In the monohydrates of P.Y.183 and P.Y.191 the water molecule is bonded so tightly that the structures are stable to about 250 °C without loss of water. Above 250 °C they slowly release water, but on cooling to room temperature the monohydrates are formed again. P.Y.183 and P.Y.191 are Ca2+ salts of organic sulfonic acids. They are even more stable than their inorganic counterpart, calcium sulfate: CaSO4·2H2O (gypsum) and CaSO4·½H2O (hemihydrate) lose all their water molecules below 160 °C. The dehydration temperature of P.Y.183 and P.Y.191 is almost 100 °C higher. This explains why P.Y.183 and P.Y.191 are suitable for plastics application, although they are hydrates [58–59].
It has been stated that, in general, the number of crystal phases known for a given compound is proportional to the time and money spent in research on that compound [60]. This is valid for organic pigments, too. For example, P.R.53:2 has been known since 1902. For 95 years no polymorphs were known; because its commercial importance was low, nobody had investigated the polymorphic behaviour. In 1997, however, a polymorph screening found 14 new crystal phases within a few months [61–63].
Typically, pigments can exist in two to four polymorphs. The record for the maximum number of observed polymorphs for an organic pigment (without counting hydrates or solvates) is held by copper phthalocyanine, which exists in at least ten, probably eleven polymorphs [64, 65]. Including solvates and hydrates, the highest number of crystal phases appears to be known for P.R.53:2, which has at least 15 crystal phases [63].
There are different numbering schemes for polymorphic forms. Some inventors use Roman numbers, but most inventors use Greek letters. The lettering generally follows the order of detection and does not correspond to the stability of the individual polymorphs. For example, the most stable phase of P.Y.12 is the α-phase, for P.B.15 it is the β-phase and for P.V.19 the γ-phase. Variations in crystal structure, for example due to the formation of mixed crystals, are sometimes denoted with primes or with superscripts. In the literature, the phase names are not always consistent, for example the γ-phase of one author may correspond to the δ- or ɛ-phase of another author. Chaos is found with P.V.19: at least 15 polymorphs have been described (α, β, BI, γ, γ′, γI, γII, γIII, γIV, several δ, Δ, ɛ, another ɛ, ζ), but only four phases actually exist (αI, αII, β, γ) [51].
In most cases, all polymorphic forms are kinetically stable at room temperature.
There are various approaches to the synthesis of a defined target polymorphic form. The following parameters largely determine the outcome:
- for hydrazone pigments: diazotization and coupling methods, direct or indirect;
- thermal conditions during synthesis and after-treatment;
- pH during synthesis and aftertreatment;
- addition of a surface active agent or resin;
- choice and timing of surface active agent;
- presence of solvents during synthesis or finishing;
- choice of solvent;
- method of precipitation or reprecipitation;
- drying process and temperature;
- milling method;
- mixed coupling (hydrazone pigments);
- chemical modification.
The extent to which each of these factors controls the formation of a particular target crystal structure depends on the individual case.
It is even possible for one batch of pigment to consist of a mixture of two different polymorphs in varying amounts. These cases should be strictly avoided in pigment production, because the application properties of the pigment will most likely vary from batch to batch. The relative amount of the individual polymorphs may strongly depend on subtle factors like temperature and concentration gradients.
Usually, thermal treatment increases the particle size, reduces the tinctorial strength and may improve the hiding power of a pigment. Reversal of this process can happen in a thermal treatment, if the polymorphic form changes: the particle size may be reduced and tinctorial strength and possibly also transparency are enhanced.
Transformations between polymorphic forms of organic pigments are strongly kinetically hindered, due to the dense molecular packing and the high lattice energy. Most transformations require elevated temperatures. Additives may help or may hinder phase transitions. In most cases the phase transformations are irreversible, and less stable phases transform into more stable one. If a phase change occurs by heating, subsequent cooling does generally not lead back to the original phase.
Polymorphic transformations can be achieved by various methods, including:
- heating the pigment as a suspension in water or an organic solvent, also under pressure;
- recrystallization from large amounts of organic solvents;
- protonation/deprotonation (especially for polycyclic pigments);
- treatment with supercritical CO2 [66];
- heating in dry state;
- kneading or milling, with or without adding organic solvents;
- variation of humidity (for hydrates).
1.5.3.3 Identification of Polymorphic Forms by X-Ray Powder Diffraction
Polymorphic forms are characterized by their X-ray powder diffractograms. Other methods like solid-state NMR or IR spectroscopy are less sensitive and may lead to misinterpretation. Powder diffractograms are measured with monochromatic radiation (usually Cu-Kα1). The measurement must be made in transmission geometry with samples filled in capillaries or placed between polymer films. Measurements on flat plate sample holders in reflection mode lead to preferred orientation and texture effects, which may strongly falsify the diffraction patterns.
Isomorphic (also called isostructural) phases are pigments that differ slightly in their chemical formula but show a similar arrangement of molecules in the solid state. In the X-ray powder diagram they show almost equal diffraction angles and reflection intensities. Isomorphous pigments are frequently more uniform in their physical and application characteristics than is true for polymorphous pigments.
X-Ray powder diagrams allow for detection of all crystalline phases. Amorphous compounds, like resins, cause an enhanced background, but no sharp reflections. Crystalline inorganic byproducts, like NaCl, CaCO3 or BaSO4, are visible as sharp peaks at higher diffraction angles.
The position of the reflections in the X-ray powder diffractogram corresponds to the lattice parameters. The reflection intensities depend on the atom types and the atomic positions within the unit cell.
In an accurately measured X-ray powder diagram, the peak widths correspond to the domain size (crystallite size), that is, the size of the homogenously diffracting regions of the particles (Section 1.5.4).
Organic pigments tend to form solid solutions (‘mixed-crystal’ phases), in which different molecules are incorporated into a common crystal lattice. Solid solutions can by distinguished from a physical mixture of the individual compounds using X-ray powder diagrams. A physical mixture results in a superposition of the diagrams of the individual compounds, whereas a solid solution causes a single pattern with shifted peak positions and modified intensities. Similarly, the incorporation of byproducts into the crystal lattice may also cause peaks shifts and changes of peak intensities.
1.5.3.4 Crystal Structure Determination
Most elegantly, crystal structures of organic pigments can be determined by single-crystal X-ray structure analysis [67], if suitable single crystals can be grown. The minimum required crystal size is 10–100 µm in all directions. Owing to the low solubility of all pigments, even at elevated temperature, the growth of single crystals from solution is usually difficult or even impossible. Especially, high-performance (hydrazone or polycyclic) pigments with good application properties, and correspondingly very low solubilities, frequently resist all attempts to grow single crystals. In some cases, single crystals of polycyclic compounds (and even hydrazone pigments) could be grown by vacuum sublimation [68].
Nowadays, there is an alternative to the single-crystal method. Crystal structures of organic compounds, including pigments, can be determined from X-ray powder diffractograms. Two approaches are used:
- If the powder is well crystalline, the X-ray powder pattern can be indexed, that is, the lattice parameters can be determined from the peak positions. Subsequently, ‘real-space methods’ are applied, in which the molecules are translated and rotated within the unit cell, until the calculated X-ray pattern matches the experimental one. With this approach, the crystal structures, for example, of a series of benzimidazolone pigments could be determined [69].
- If the indexing fails, possible crystal structures can be calculated by global lattice energy minimization procedures, either using force fields, or using quantum-mechanical methods (see below). To identify which of the calculated possible structures corresponds to the actual polymorph, powder patterns are calculated for all resulting low-energy structures, and compared with the experimental patterns. This approach was applied to solve the structure of a methyl derivative of P.B.80 from a powder pattern with 12 peaks only (Figure 1.18) [70]. The method can also be used for indexed or partially indexed X-ray powder diagrams [71–73].

Figure 1.18 Structure determination from X-ray powder data of the methyl derivative of P.B.80. Top: Experimental X-ray powder diagram. Bottom: Simulated diagram of the predicted crystal structure. The difference between the two diagrams is as small as for a rotation of the molecule by 1°. The crystallinity of this sample represents the lower boundary required for structure determination from X-ray powder data. The crystal domain size is about 20 nm.
In all cases the crystal structure is finally fitted against the full powder diffractogram (Rietveld refinement, see Figure 1.19). The use of synchrotron data instead of laboratory data generally increases the accuracy of the obtained structure, if the powder is of good crystallinity.

Figure 1.19 Rietveld refinement of P.R.57:1 (β-phase) [74]. The experimental X-ray powder diagram (laboratory data, measured in transmission mode) is drawn as black points, the calculated diagram as red line. The difference curve is shown in blue. The vertical tick marks indicate the reflection positions. At 36°, the scale changes by a factor of 10.
An alternative to X-ray methods is electron diffraction [75]. A powder containing crystals with sizes far below 1 µm is fully sufficient; however, data recording, structure solution and refinement are much more challenging than for X-ray diffraction. Nevertheless, the crystal structure of the ζ-phase of P.R.53:2 could be solved this way [75].
For difficult cases a combination of electron diffraction, X-ray powder diffraction and lattice-energy minimization is recommended [52].
Some pigments form amorphous phases, in which the molecules are either arranged in a random way or exhibit a local ordering only.
Structural investigations on nanocrystalline and amorphous pigments can be made by analysing the pair-distribution function (PDF). The PDF, also called ‘radial distribution function’, G(r) represents the probability G of finding two atoms with an interatomic distance r. It is weighted with the scattering power of the corresponding atoms and summed over all atom–atom pairs. The PDF is derived from a carefully measured X-ray powder diagram, and provides information on the local structure. It can be used, for example, to identify polymorphs in crude, unfinished or severely milled pigments. The PDF is also applied to determine whether an amorphous phase actually contains a fully random arrangement of molecules or if there is still a local ordering of neighbouring molecules, with the corresponding effect on the optical properties of the pigment.
1.5.3.5 Prediction of Crystal Structures; Crystal Engineering
Possible crystal structures of organic pigments can be predicted by lattice-energy minimizations.
Two main methods are used, force fields and quantum mechanical methods. Force-field methods require much less calculation effort. Their accuracy depends on the applied force-field parameter set and on the applied atomic charges. The best results are obtained with tailor-made force fields, where all parameters are fitted to quantum-mechanical calculations for each molecule individually. Quantum-mechanical methods such as DFT-d (density functional theory with dispersion correction) give very accurate results [76].
The calculation time is drastically reduced if the unit cell parameters can be used as input, which may be known from an indexed powder diagram, or from electron diffraction.
A fully theoretical prediction of possible crystal structures without reference to experimental data starts from a set of thousands of crystal structures with random lattice parameters, random position and orientation of molecules in all frequent space groups. For flexible molecules, the intramolecular degrees of freedom have to be included, too. All structures are optimized. The resulting structures are sorted according to energy. Duplicates are removed. The lowest-energy structures represent predicted possible polymorphs of the given compound [77].
Based on the predicted possible crystal structure, properties of the pigments can be deduced, like hydrogen bond pattern, density, colour, crystal morphology, surface polarity and so on. This may be helpful especially in those cases where synthesis is difficult, or when the outcome of an attempted synthesis is questionable.
Crystal structure predictions can also be used to investigate whether a compound is likely to have additional, hitherto unknown polymorphs. In a crystal structure prediction of P.Y.74 only one low-energy structure was found. This structure corresponds to the experimental structure, which suggests that there is no other polymorphic form. The prediction was comfirmed experimentally: no other polymorphic forms were found in an extensive polymorph screening [78].
Crystal engineering is the design and synthesis of new materials with targeted properties. It is based on knowledge of the crystal structure, either from experiments or from crystal structure predictions [65].
Crystal engineering was successfully applied on P.R.170. The crystal structure of the commercial γ-phase was determined by lattice-energy minimizations with the help of a partially indexed X-ray powder diagram [72]. A thorough inspection of the crystal structure revealed that the structure contains a small void close to the ortho-position of the diazo compound (Figure 1.20). Lattice-energy minimizations suggested that the lattice energy would improve if this void is filled by an additional substituent in the ortho-position of the diazo compound. This substitution pattern has never been tried before, because synthesis of the diazo compound requires additional effort. Introduction of a chlorine atom led to a different molecular packing with an undesired orange shade and worse properties. Obviously, the chlorine atom is larger than the void. However, it turned out that 10–20% of the chlorinated molecules can be incorporated into the lattice of γ-P.R.170 without changing the molecular packing. The resulting solid solution led to the desired red pigment with improved light fastness [72, 79].

Figure 1.20 Crystal engineering on the γ-phase of P.R.170. The small void marked by X can partially be filled by an additional substituent, leading to improved application properties.
1.5.4 Crystallinity
Most pigments, such as members of the hydrazone series, emerge from the manufacturing process as fine powders of poor crystallinity. Drying of the crude presscake immediately after synthesis without further finishing usually results in considerable agglomeration of the fine particles, which may later resist even the most determined dispersion efforts.
Considering the crystal imperfections that are typically found in all crystals, the crystal quality of organic pigments is a major concern. The external surface of any crystal exhibits several defects, which expose portions of the crystal surface to the surrounding molecules. Impurities and voids permeate the entire interior structure of the crystal. Stress, brought about by factors such as applied shear, may distort the lattice and modify the lattice parameters. It is also possible for the three-dimensional order to be disturbed by various types of lattice defects like dislocations (e.g. line dislocations or screw dislocations), stacking faults, small-angle grain boundaries, orientational disorder of molecules or complete chains, inclusion of starting material or byproducts and so on.
The crystallinity is determined by X-ray powder diffraction. In an accurately measured X-ray powder diagram, the peak widths correspond to the domain size (crystallite size), that is, the size of the homogenously diffracting regions of the particles. The domain size depends on the particle size and on the crystallinity, that is, on the number and nature of lattice defects. The peak widths increase with decreasing domain sizes. The particle dimensions as well as the crystallinity may be strongly anisotropic, that is, different in the individual lattice directions, which may cause variable peak widths within the same powder pattern.
The crystal domain size can be calculated according to the Scherrer formula:
in which:
| L | = | mean domain size |
| k | = | shape factor (approx. 0.9) |
| λ | = | wavelength |
| b | = | full peak width at half-maximum intensity in radians |
| θ | = | diffraction angle |
Note that the peak width and the diffraction angle must be given as θ, not as 2θ.
Depending on the X-ray powder diffractometer, the upper limit for the detection of peak broadening is of the order of 500 nm. Above this value the line width is determined by the instrument only. The lower limit is of the order of 10–20 nm. Crystallites with even smaller sizes result in very broad, overlapping peaks, which impede reliable determination of the peak widths.
Untreated pigment samples are frequently nanocrystalline. Nanocrystallinity is a state between amorphous and crystalline. We define pigments as nanocrystalline, having domain sizes below about 20 nm, which roughly corresponds to 10–20 unit cells in each direction.
There is no sharp boundary between ‘crystalline’, ‘nanocrystalline’ and ‘amorphous’. Usually, a pigment is regarded as ‘crystalline’ if the X-ray powder diagram contains sharp reflections, as ‘nanocrystalline’ if the X-ray powder diagram consists of broad reflections, and as ‘amorphous’ if there are only some broad humps, but no reflections. However, the absence of reflections does not mean that there is no preferred arrangement of molecules in the solid state. A typical small crystallite with a size of 5 × 10 × 20 nm, containing 10 × 10 × 10 unit cells with 4000 molecules, would cause only humps in the X-ray powder diagram, although the molecules form an ordered arrangement. The same holds for larger crystals, which contain so many defects that the individual domains are too small to give sharp reflections in the X-ray powder pattern.
Lattice imperfections and voids ‘heal out’ during the finishing that commonly follows pigment synthesis. The crystallinity of a pigment is thus improved by thermal treatment. Figures 1.21 and 1.22 show examples of pigments before and after finishing or recrystallization. In solvent grinding or salt milling, two contrary effects may play a role: Owing to the mechanical forces the particles become smaller and the number of lattice defects increases, resulting in a worse crystallinity and a smaller domain size. On the other hand, the presence of a solvent and the elevated temperature during grinding or milling may cause a healing of the lattice defects and a recrystallization of the pigment, leading to an improved crystallinity and an increased domain size.

Figure 1.21 X-ray powder diagrams of P.Y.83 showing different crystallinities and particle sizes. Top: Transparent, unfinished pigment with small domain sizes, used for printing applications. Bottom: Finished pigment with better crystallinity and larger particle sizes, showing a more orange shade, a higher opacity and better fastness properties, suitable for automotive finishes. Both diagrams were measured under the same conditions with Cu-Kα1 radiation in transmission geometry on a Stoe Stadi-P diffractometer.

Figure 1.22 X-ray powder diagrams of P.Y.14 showing different crystallinities and particle sizes. Top: Transparent, unfinished pigment with small domain sizes, used for printing applications. Bottom: Well crystalline pigment recrystallized from boiling dichlorobenzene (non-commercial). Both diagrams were measured with Cu-Kα1 radiation in transmission geometry on a Stoe Stadi-P diffractometer.
The properties of organic pigments are closely related to the quality of their crystals. With decreasing crystallinity, that is, with decreasing domain sizes and increasing number of lattice defects, the fastness properties of pigments like light fastness, weather fastness and migration fastness get worse. The effect is of an energetic nature: the more favourable lattice energy of an undisturbed crystal lattice enhances its fastness properties. Additionally, more perfect crystals tend to have a lower surface-to-volume ratio, which reduces the solubility and decreases the possibility of chemical reactions (e.g. with radicals) on the surface. X-Ray powder diffraction provides the instrumentation that is necessary to monitor the crystallinity as an indication of application properties during synthesis and, even more important, during finishing.
A few pigments, for example, the triaryl carbonium compound P.B.56, are sold as amorphous compounds.
1.6 Important Application Properties and Concepts
The key application properties of organic pigments consist of all properties of the pigmented system that are determined, or at least strongly influenced, by organic pigments. These include, first of all, the fastnesses, that is, stability to a wide variety of effects such as light, weather, heat, and organic solvents. Pigment-related problems that arise in the processing or use of the pigmented medium, such as plate-out, chalking, flocking and so forth, fall into this category. The dispersibility of the pigment and the rheological properties of the pigmented medium are particularly important. Colouristic properties and concepts, such as colour depth, tinctorial strength, hue, covering or hiding power and transparency, are often discussed separately from application properties.
1.6.1 Colouristic Properties (by F. Gläser)
The colouristic qualities of a pigment have to do, broadly, with the description of its colour-imparting action, that is, the characterization of the colour nuance produced through its use, as well as its effectiveness, covering power and transparency. Obviously, these are fundamental properties that govern the industrial use of a pigment and its economic value.
The colouristic qualities of a pigment and of its colour are not evaluated in the as-delivered condition but always after it has been processed in a way corresponding to the application. Information gained for one application cannot, however, be directly extended to other application processes.
Because colouristic assessments are essentially judgments of colour effects, colouristic practice long rested solely on the colourist's trained eye. Today, the measurement of colour is a mature field of science, and colourists employ theories of the optical behaviour of pigmented layers.
This section will discuss some important concepts from colouristic practice and the optical properties of pigmented systems. Space considerations permit treatment of only the most vital concepts. The reader must consult the literature for further details and accounts of special problems [80, 81]. A review on the effect of crystal structure on colour application properties has been published [50].
1.6.1.1 Colour
The colour of an object is a sensory impression received by the individual and triggered by a colour stimulus. The colour stimulus consists of light from the object incident on the eye, ‘light’ denoting electromagnetic radiation in the range of wavelengths between about 400 and about 700 nm. In terms of the colour stimulus, the colour impression depends on the distribution of radiant energy over the wavelengths of the visible spectrum. For a given colour stimulus, however, the perceived colour also depends on the observer's individual ability, the observer's instantaneous adaptation to illumination conditions, and other colours perceived in the visual field along with the object.
For non-self-luminous objects, the colour stimulus is determined first by the illumination and second by the optical properties of the object viewed. The effect of illumination is due to differences in the distribution of light energy within the visible part of the spectrum. With respect to the object, it is important which fraction of the light incident on it reaches the observer's eye. This fraction is generally a function of the wavelength, and the nature of this dependence determines the colour. Both the fraction and its wavelength dependence can be controlled through the type of pigment and the level of pigmentation.
To describe a colour impression unambiguously, three independent colour properties must be measured. In colouristic practice with visual evaluation, these are usually (i) the hue, which describes whether the specimen is, say, red, blue, yellow, or green; (ii) the colour depth, which tells how strongly coloured it is; and (iii) the cleanness, which characterizes (roughly speaking) the brilliance of a colour. Other quantities could be selected for such a description, such as lightness or chroma. The lightness of a colour is given by the grey equivalent to it; chroma describes how far a colour departs from achromatic ‘colours’ (white, grey, black). Because the verbal description of colour perceptions is less precise, real specimens, for example, coloured chips and the description of colours by refering to such specimens, play a significant role.
The industrially used colorimetric systems are standardized by the Commission internationale de l'éclairage (‘CIE, in English International Commission on Illumination’). One method of characterizing a colour is to state the CIE ‘tristimulus values’ X, Y, Z, which are calculated with the formulas:
The use of these formulas involves subdividing the visible spectrum into L (at least 16) intervals. The subscript n labels these wavelength intervals. Jn characterizes the intensity of illumination in interval n, while Rn is the average reflection factor for this interval. The quantities xn, yn and zn are found in tables (contained in standards) for typical illumination conditions and for an observer with ‘normal’ vision. If two coloured samples are of the same colour, their X, Y and Z values are equal pairwise. If two objects have the same colour under a given illumination, this does not necessarily imply that they have the same colour under all other illuminations. This can be so only if all the Rn values for the two objects are equal in pairs. If the colours of two objects match only under certain illumination conditions, the objects are said to have metameric colours.
Colours specified in terms of the tristimulus values X, Y and Z are fairly hard to visualize. For this and other reasons, various different colour systems have been devised. The colour coordinates in these systems can be calculated from the X, Y and Z values, which are of central importance to colour measurement because of their close link to measurable quantities.
For example, the variables:
are frequently used. These make it possible to visualize relationships between distinct colours provided the effects of lightness can be neglected. Figure 1.23 presents the CIE chromaticity diagram from DIN 5033, showing the approximate location of the lightest colour for any given pair (x, y). (The abbreviation ‘DIN’ stands for ‘Deutsches Institut für Normung’. i.e. German Institute of Standardization).

Figure 1.23 CIE chromaticity diagram from DIN 5033 (showing ‘white point’ E).
The CIELAB system (‘L*a*b* system of the Commission internationale de l'éclairage’) [82] uses the rectangular coordinates L*, a* and b* or the polar coordinates L*, c*, h. The latter show a good correlation to the quantities lightness, chroma and hue. L* denotes the lightness, with values of L* = 0 for black, and L* = 100 for white. The chroma c* is zero for white, grey or black; the maximum achievable value (pure color) depends on the hue. The hue angle h adopts values of h = 0 for red, h = 90 for yellow, h = 180 for green and h = 270 for blue (Figure 1.24) [83]. The L*a*b* values can be transformed into the L*c*h values by
The values for L* are identical in both systems.
Figure 1.24 Characterization of colours in the CIELAB system.
1.6.1.2 Colour Depth
While concepts like hue and purity easily lend themselves to verbal description, major difficulties arise in any attempt to discuss ‘colour depth’ or ‘depth of shade’. The only way to deal with this concept (as with other aspects of colour) is to create specimens having the properties discussed. For this reason, a collection of samples equal in colour depth was prepared around 1935 so that the fastnesses of various dyes could be meaningfully tested [86]. Studies by Raabe and Koch [84] and by Schmelzer [85] suggest that ‘equal difference (or distance) from white’ is an essential criterion in selecting specimens of equal colour depth. Some colour-depth levels are set apart as ‘standard depths of shade’ (SD) [85]. These find use particularly in the preparation of specimens for fastness testing; they permit a consistent way for the presentation of different colourants and describing their effectiveness under various service conditions.
The colour depth can be measured colourimetrically by the methods of Gall and Riedel [86]; it is expressed in terms of the deviation from one of the standard depths of shade. In a modification of this system, proposed by Schmelzer [85], the colour depth is rated on a continuous scale from 0 to 10. Because it is a simple matter to assign a colour depth to a specimen by colourimetry, this concept has come increasingly into use.
1.6.1.3 Colour Differences
The assessment of colour differences [89, 90] is at least as important as that of individual colours. There are nearly always differences between similar colours. If specimens are viewed next to each other, even very small colour differences can be detected visually. Given sufficient experience and a standard system of notation, visual judgments are largely consistent from observer to observer.
In colourimetric terms, colour differences are generally characterized by the distance between two colours in one of the colour notation systems, most commonly the CIELAB system. The difference determined in this way represents the ‘total’ colour difference. Since a colour is characterized by three quantities, a colour difference can also be expressed in terms of three difference components (i.e. differences in the colour coordinates) and thus described in greater detail. Visually equal colour differences between almost equal colours may, upon colourimetric measurement, be considered distinct if the colours of one pair differ strongly from those of the other pair. The formulas for colour differences are further elaborated to remedy this defect.
1.6.1.4 Optical Behaviour of Pigmented Coatings
Physically, a pigment imparts colour by influencing light propagation in the pigmented system. Pigments attenuate light by absorption (by transforming radiant energy into heat), and/or alter the direction of propagation through scattering and/or reflection.
The way in which the propagation direction is changed depends on the size of the pigment relative to the wavelength, the difference in refractive index between the pigment and the vehicle and also on the direction of incidence. If the pigment dimensions are large in comparison with the wavelength, reflection and refraction are described by the laws of geometrical optics. If they are comparable with the wavelength, light is scattered in various directions; this scattering depends in a complex fashion on the dimensions, the relative refractive index and the absorbing ability of the pigment. If the pigment has very small dimensions compared with the wavelength, the direction of propagation is not altered; the pigment works entirely by absorption.
As a model of a pigmented system, consider a pigmented coating on a flat substrate, with the whole illuminated and observed from the coating side. A great number of individual effects can occur in this situation, as Figure 1.25 shows (except for refraction at the surface of the coating).

Figure 1.25 Reflection, scattering, and absorption of light in pigmented coatings.
How much these individual processes contribute to the overall behaviour of the system will depend on several factors: the vehicle, pigment, substrate, and pigment concentration. What is more, such processes usually depend strongly on the wavelength, so that drastic changes in response to wavelength can occur particularly with organic pigments. There is no general quantitative description of such a complex system, at least not in a practically usable form.
A quantitative description does become possible, however, if the system under examination satisfies special conditions. These include diffuse, monochromatic illumination, homogeneous pigmentation, isotropic scattering in the coating, no difference in refractive index between vehicle and air, and a coating so thick that the substrate has no effect on the exiting radiation. This is the special case treated by the Kubelka–Munk theory.
In this model, the relationship between the fraction R of incident light that exits the coating (the remission) and the constants K and S characterizing absorption and scattering, respectively, is given by the formula:
If K and S are known, R can be calculated. In the case of several pigments together, the constants K and S can be calculated with the formulas [87]:
with:
| Kn, sn | = | absorption and scattering constants per unit concentration for pigment number n; |
| cn | = | concentration of pigment n; |
| KB, SB | = | absorption and scattering constants of the vehicle (bulk phase of the coating); |
| N | = | number of pigments present. |
In a given vehicle, kn, sn, KB and SB can be determined by calibration against reference systems. Thus, K and S and then R, can be calculated. If this is done for all wavelengths needed in the computation of X, Y and Z values, then X, Y, Z, and thereby the colour expected for the pigmented system, can be calculated.
Many of the stated restrictions can be lifted (at the cost of using more complicated formulas) when these conditions are not adequately met. For other systems, it may be more appropriate to use approximations based on assumptions different from those in the model described above, which applies to systems with approximately optimum covering power. Such an approximation has been presented by Hoffmann [88] and simplified by Schmelzer. It is employed for highly transparent coatings where the substrate plays a significant role; specifically, it describes the behaviour of printing inks.
These calculations are important not solely for understanding the behaviour of pigments. They also provide a basis for calculating formulations to match given colour samples, for determining the colour imparted by a single pigment at a desired concentration (given the effect of a different concentration in the system) and for solving many other problems.
1.6.1.5 Tinctorial Strength
Like other optical characteristics, the tinctorial strength of a pigment, that is, its ability to add colour to a substrate, is controlled by the conditions under which it is applied. Tinctorial strength may be defined either as an absolute value or relative to another pigment. Attempts to assess absolute values by visual methods at best afford semiquantitative results; while the relative strength of one pigment compared to another is more easily assessed by a human observer. It is determined quantitatively by comparing a series of samples featuring varying amounts of a pigment incorporated in a substrate with a corresponding series of pigment/substrate systems containing the reference pigment. Test and reference samples are then compared and matched as far as possible. The ratio between the known pigment concentration of the respective test sample of a matching pair and the pigment concentration of the corresponding reference sample is used as a measure of the relative tinctorial strengths of the two pigments.
If no exactly matching sample can be found, the concentration that might lead to a match is approximated through visual interpolation from the two nearest reference samples. If it is impossible to arrive at an exact match for any concentration, then the concentration is found which experience shows to be equivalent. The corresponding tinctorial strength is supplemented by a verbal description of the remaining differences in hue and cleanness.
Colourimetrically, the absolute tinctorial strength of a pigment in a vehicle system is best described by referring to the depth of shade that is achieved under specific conditions of sample preparation. It is possible to either measure the amount of pigment that is necessary to produce a certain depth of shade or to refer to the depth of shade that can be produced with a certain amount of pigment. The second approach, which is based on a defined amount of pigment, appears particularly convenient for the user who is interested in comparing the maximum depth of shade that can be achieved under different working conditions. Such values are illustratively plotted by using the continuous depth of shade scale designed by H. Schmelzer [85].
Other approaches to determine absolute tinctorial strength in a sample are based on the assumption that the tinctorial strength of a pigment is defined by its light absorbing properties. The absorption coefficient for a known concentration is usually measured at the wavelength of maximum absorption; an alternative method of determining the tinctorial strength involves summarizing the absorption coefficients over the entire range of the visible spectrum. In the latter case, absorption coefficients are sometimes weighted using different weights for each wavelength. Absorption methods may occasionally present problems, particularly in trying to compare pigments that are very different in shade. Depending on the approach, large parts of the spectrum might simply be ignored (if only the absorption at the absorption maximum is used), or they are represented somewhat indiscriminately (if the sum of absorption coefficients is used). However, those parts of the spectrum that are not correctly included in the calculations obviously contribute to the perception of colour by the human eye. Weighting the absorption coefficients according to their wavelength does not entirely alleviate the problem, because the correct choice of weights is not always obvious.
The relative tinctorial strength is defined in relation to a standard rather than being an absolute property of a pigment. For colourimetric assessment, the absolute tinctorial strength of a sample pigment and that of a reference pigment are determined by the same method and the ratio between two such absolute values is then calculated. Despite the variety of options, however, both the absorption at maximum wavelength and the absorption sum over the visible range of the spectrum remain the most common criteria for the determination of the relative tinctorial strength.
It is also possible to find the amount of pigment necessary to produce a match of a reference sample containing a known amount of pigment. The relative tinctorial strength is then defined as the ratio between the two amounts. In this case, two samples are already considered to match if they exhibit the same depth of shade; an exact match is not always found.
After all differences in tinctorial strength are accounted for, any remaining deviation is due to the inherent difference in shade between the two pigments and cannot be eliminated by simply adapting the amounts. It is useful to describe the remaining colour difference by referring to the CIELAB system.
Quantitative comparison of numerical values from several different sources requires some caution, because the tinctorial strength of a given pigment is largely defined by the working conditions, which govern its application, as well as by the methods of determination and evaluation.
1.6.1.6 Hiding Power
In discussing the performance of a pigment within a vehicle system, it should not be forgotten that there are certain aspects that relate to the substrate onto which it is applied. The hiding power of a pigment, for instance, refers to the ability of a layer of pigmented medium to conceal any differences in substrate colouration so that they become invisible. It is defined as the area over which a certain amount of pigmented paint can be spread without losing its opacity. It is also possible to refer to the minimum thickness of layer that is necessary to conceal a substrate.
A layer that effectively conceals its substrate must be scattering. The necessary amount of scattering depends on the thickness of the layer, the absorption of light within the layer, and the magnitude of the colour differences of the substrate. The less absorbing the paint or the larger the colour differences of the substrate, the higher the scattering power needed to adequately hide the substrate. The hiding power depends also on the wavelength of the incident radiation. The hiding power of a chromatic pigment can be different for different colours of the substrate [91].
The hiding power of a pigmented layer is usually determined by applying it to a substrate with differently coloured patches. Black stripes on a white background or a black/white or grey/brown chequerboard surface with a standardized reflectance (e.g. R80% for white and R5% for black) are commonly used. To quantify the hiding power of a layer in relation to a standardized substrate, the maximum area may be specified over which the test paint can be spread without losing its opacity. Alternatively, it is also possible to find the minimum thickness of layer that just effectively hides to the eye any difference in colour between the differently coloured patches of substrate surface. By definition, this is the point at which the colour difference equals one in the CIELAB system.
According to ASTM D 2805-70, the hiding power of a layer is characterized by the ratio of the Y values (as defined in the CIE colour system) over a black and a white patch of substrate. If this ratio is larger than 0.98, the layer is referred to as hiding. This criterion occasionally fails if applied to brightly coloured pigments.
1.6.1.7 Transparency
The list of system-dependent parameters includes the transparency of a pigmented layer. It is usually determined by applying the pigmented system to a black background whose darkness is retained or reduced according to the transparency of the layer. Scattering increases the opacity of a layer. Quantitative evaluation relies on the difference in colour between the pigment–vehicle system spread over a black surface and a black surface that is covered with pure vehicle only. The amount of pigment that is necessary to create a difference in colouration of Δ E = 1 using the CIELAB system for colour differences provides an indication of pigment transparency.
1.6.2 Fastness to Solvents and Special Application Fastness
1.6.2.1 Organic Solvents
Although, according to definition, the ideal pigment is practically insoluble in its medium of application, organic pigments may in reality deviate more or less from this postulate of insolubility, depending on the medium in which they are applied and the conditions under which they are processed. Since a pigment that is to a certain extent soluble in its carrier is expected to perform poorly and may even recrystallize, bleed or bloom, it is important to prevent pigment dissolution. Factors that control the solubility of a pigment in its vehicle include the choice of solvent, chemical structure, particle size (Section 1.7.7) and temperature. To demonstrate the correlation between solubility and temperature in organic pigment/solvent systems, Figure 1.26 traces the solubility of P.Y.1 and P.R.3 in dibutyl phthalate at temperatures between 25 and 90 °C [14].

Figure 1.26 Relation of the solubility of Pigment Yellow 1 and Pigment Red 3 in dibutyl phthalate to the temperature.
There are certain accepted tests used to determine the extent to which a given organic pigment tolerates solvents. Experimental procedures commonly involve enclosing a certain amount of pigment powder into a piece of fluted filter paper, which is inserted into a test tube containing the organic solvent. The sample is then kept in the organic solvent for 24 h at room temperature, after which the results are evaluated. The extent to which the solvent is coloured indicates the fastness of the pigment to this particular solvent. Although laboratory studies cannot reliably imitate a commercial environment, the analytical results do allow general conclusions and assist in selecting the suitable pigment for a particular application. In this stability test, the tendency of a pigment to lose colour in a certain medium, for instance, is usually associated with reduced fastness to overspraying.
The way in which a pigment behaves toward a specific solvent has a considerable impact on how it can be employed by the user. If a pigment that is partially soluble in a solvent is processed in a system containing that solvent, then recrystallization may occur. This in turn alters the colouristic, rheological and fastness properties of the product.
Several potential users, such as the printing industry, are interested in the solvent fastness not only of a pigment but of an entire pigment–vehicle system. Standardized tests are available. A proof print of a certain size is placed inside a test tube and allowed to remain in the target solvent at 20 °C for 5 min. The change in solvent colour is determined and the print dried and compared with an untreated specimen. Standard solvents [92] are ethanol or the following mixture:
- 60 vol% ethanol,
- 30 vol% ethyl acetate,
- 10 vol% 1-methoxy-propanol-2.
However, the results do not necessarily reflect the fastness to overlacquering in actual application, because overprint varnishes may contain other solvents as well. Besides, a series of additional factors such as the effect of plasticizers, uneven flow of the pigmented lacquer, and so on, play a role in pigment performance. The fastness to overlacquering is therefore most realistically determined in actual application.
Pigment performance also includes fastness to transparent lacquer coatings (‘silver lacquer’), that is, fastness to transparent enamels that are applied to metal deco prints to give them rub and scratch fastness.
1.6.2.2 Water, Soap, Alkali and Acids
The test methods discussed in this context are broad in scope and relate to various applications. In some such tests, a pigment powder is extracted with water and its contents analysed; other procedures reveal the stability of a pigmented system with regard to water, acids or bases. Several of these tests have developed into industrial standards.
Various techniques are used to determine the water-soluble content of a pigment.
The cold extraction method [93] involves moistening defined amounts of pigment powder (between 2 and 20 g) with small amounts of water, alcohol or a suitable wetting agent. Freshly distilled or completely deionized water (200 ml) is then added and the sample allowed to remain in this solvent for 1 h at ambient temperature. After thorough shaking, the liquid is removed by filtration.
The hot extraction method is a variation of this procedure. The aqueous pigment suspension, prepared as above, is boiled under reflux for a certain time, usually 5 min, cooled rapidly, and filtered. A known amount of extract is then dried by evaporation and the weight of the residue determined, or the extracted pigment weighed and the dissolved portion determined by calculating the difference.
The acid or alkali number of a pigment is determined by titrating the aqueous extract of 100 g of pigment with a 0.1 N alkali (acid) solution [94].
Whenever organic pigments are analysed, it is not uncommon for the results of the two extraction methods to deviate considerably. Depending on the pH of the medium in which the synthesis is carried out, such as the coupling slurry for hydrazone pigments, occlusion or adsorption effects may trap portions of the soluble substances inside or on the agglomerates. It is impossible to remove such impurities by pigment filtration and intensive washing, and even the effect of hot extraction procedures tends to be slow and unsatisfactory. Considerable amounts of such soluble species may remain within the pigment even after hours of refluxing and repeated filtration with freshly distilled water. A pH or conductivity analysis of the extracts will readily reveal this unsatisfactory condition.
It is also possible to determine the pH of the aqueous pigment extract from either the cold or the hot process. A frequently used modification of this method involves measuring the pH of the pigment slurry with a glass electrode directly after reflux [95], without prior filtration. There are certain disadvantages to this technique if it is used on pigments whose surface is covered with resin or has been treated with other additives such as amines: resins and similar materials tend to occlude portions of the surrounding medium more than do untreated pigment surfaces.
There are commonly accepted experimental standards for the determination of conductivity and specific resistance in aqueous pigment extracts [96]. The electrical conductivity (y) is calculated from the electrical conductance; its inverse is the specific resistance ξ1/γ, derived from the electrical resistance. Additional experimental methods have been developed for the determination of soluble sulfates, chlorides and nitrates [97].
From the user's point of view, the ability of a pigmented system to withstand exposure to water, alkali or acids is frequently of more importance than an evaluation of pigment extracts.
The printing industry uses standardized proof prints for all printing methods to examine the fastness of a pigment to water or alkali. The sample prints are placed between or on top of filter paper soaked with water or with a 1% sodium hydroxide solution [98]. The resulting soaked print with its filter paper is sandwiched between two glass plates, which are then placed underneath a 1 kg weight. Water fastness is evaluated after 24 h; alkali fastness tests require only 10 min. Prints or printing inks which neither migrate into the filter paper nor undergo a colouristic change are said to be water or alkali resistant.
There are no similarly defined standards to indicate pigment performance in connection with coatings; however, various methods have gained acceptance throughout the automotive finishes industry. A generally accepted technique to determine fastness to alkaline agents involves applying a 5% sodium hydroxide solution to the coating and allowing the covered specimen to remain in a circulating air chamber at 70 °C for 1 h. The purpose is to evaluate the properties and fastness of a coating under conditions that reflect those in a commercial car wash. Acid tests commonly use 1 N sulfuric acid with a certain amount of iron(II) sulfate at 70 °C ± 1 °C for 1 h. The effect on the colour of the test coating should not exceed 2 CIELAB units, and its gloss should not be reduced by more than 10%.
Although it is uncommon for organic pigments to be affected by acid or alkali, colour changes are occasionally observed with laked pigments or pigments containing free acid or amino functions. These may form salts if they are exposed to acids or bases.
The requirements of the coatings industry depend on the context in which a pigment is to be used. Exterior house paints, for instance, are expected not only to exhibit good weatherfastness but also to be fast to lime and concrete. Fastness to lime is frequently tested by colouring freshly slaked lime with the pigment dispersion or pigment preparation, applying the mixture to concrete sheets, and after 24 h comparing the colour of the test sample with that of a freshly prepared specimen. Very few organic pigments can be mixed directly with cement, but several meet the requirements for paints to be applied to concrete surfaces. The outcome of such a test is crucial, because the colour of a pigment that is not sufficiently fast to its carrier material will deteriorate or change rapidly when it is exposed to weather.
Likewise, the evaluation methods for polymer binders are designed specifically for certain target materials. One such procedure is known as the ‘tropics test’ for polyurethane coatings. The ability of a coloured test sample to resist hydrolysis is determined by exposing it for 7 days to a climate of 100% humidity at 70 °C and then extracting it with ethyl acetate; or, alternatively, by submitting the hydrolysed specimen to a crock test. Coatings may become more prone to hydrolyse due to the influence of certain pigments; if a pigmented system is used, the binder may be responsible for hydrolysis.
1.6.2.3 Pigment Performance in Special Applications
There is no precise definition for the frequently used term ‘application fastness’ of a pigment. It usually refers to the behaviour exhibited by a finished product used in accordance with the specifications. The term may thus refer to a print, a coated object or a plastic product, and the list of features ranges from properties such as lightfastness and weatherfastness to migration fastness and fastness to solvents. In this context, there is a certain amount of emphasis on features that play a role in connection with packaging materials and packed articles.
Test methods for prints are broad in scope to suit various practical purposes. Users are frequently interested in the fastness of a product to soap and detergents [97]. To determine the fastness of a print to a freshly prepared 1% aqueous solution of a special test soap, the printed sample is placed on top of several layers of filter paper soaked with the test solution. The resulting test specimen is then sandwiched between two glass plates, mounted underneath a 1 kg weight, and held at 20 °C for 3 h in a chamber saturated with water vapour. The printed sample is then removed, rinsed with distilled water and dried. The resulting colour change of the test specimen is evaluated by comparing the colour with an untreated print. Any colouring of the filter paper is noted as well.
Likewise, pigment performance in connection with detergents frequently decides the commercial range of a product. It is generally considered useful to test the target detergent directly as opposed to the more common approach of simultaneously evaluating alkali and soap sensitivity. The latter approach has a major impact on the range of available pigments because the demand for perfect fastness to soaps under the conditions of actual processing and use eliminates so many types. Because of this, numerous shades are not accessible for this use.
The requirements for prints used in the packaging industry are determined by the packaged material, which explains the wide variety of standards that govern pigment application in this area. The list includes fastness to materials such as cheese, grease, oil, paraffin, wax and certain spices [98]. Tests are carried out by placing the test print on top of the medium in question, printed side down. A weight is then placed on the sample. It is important to remember that any fastness determined by this method is only valid for the material that was actually used in the test. Spices, for instance, may act very differently, depending on their age, storage temperature and milled form.
Likewise, the behaviour of a print in connection with sausage, ham, bacon or fish, or its sensitivity to cleaning agents, disinfectants, bath soaps, essential oils or fertilizers, may be determined accordingly.
Coloured household items made of plastics and other polymers [99] are tested similarly. The effect of food may be imitated by using coconut oil or peanut oil (‘coconut oil test’). Strips of filter paper soaked with these materials typically remain in contact with the test sample for 5 h at 50 °C.
The packaging industry is particularly interested in the stability of a pigment to sterilization and to heat sealing, as well as its effect on the heat sealability of a printed material. In a sterilization test, a pigment may be exposed to hot air, steam or water. Wet tests are carried out either at ambient pressure or in an autoclave. Colour-change comparisons with reference specimens and, for wet methods, the extent to which the water is coloured are indications of the fastness of a print to sterilization. A pigment is said to be heat-sealable if under certain defined conditions appreciable cohesion is attained between printed film and substrate. The test is thus standardized with respect to temperature, time and pressure, and the test conditions are expected neither to weaken the sealing site nor to induce a colour change in the print. Sealability tests consist of both print/print adhesion and print/substrate interaction, including pigment application to coated substrates. Printed layers are heat-sealed at defined temperatures with grooved heated jaws; both time (a few seconds) and pressure are standardized. Factors that may possibly preclude pigment application in this packaging sector include colour change, physical damage to the printed film, or adhesion to the heat-sealing equipment.
Different demands are placed on a pigment that is targeted for covered or laminated printing. In this technique, a reverse impression is printed onto a plastic film and laminated by means of one or two component adhesives, laminating resins and wax onto another substrate such as paper, aluminium foil or polymer foil. These substances may adversely affect pigment performance. For high gloss applications, a normal impression is covered with a clear plastic film.
1.6.2.4 Textile Fastness Properties
The term ‘textile fastness properties’ is used in this context primarily to designate the ability of spin dyed or printed textiles to retain their colour value throughout processing, application and use. The national body active in the development of standardized tests in Germany is the Deutsche Echtheitskommission DEK (DEK = German Commission for Fastness). This body is a member of the European Fastness Convention (ECE) and is represented by the Deutscher Normenausschuss DNA (DNA = German Standards Committee), which follows the recommendations of the International Standards Organization (ISO).
To determine the colourfastness of a dye or a pigment, it is useful to maintain a constant depth of shade (Section 1.6.1.1), usually 1/1 SD (SD = Standard Depth of Shade). The tendency of a sample of spin dyed or printed textile material to change colour may either be determined on an individual swatch of fabric or, if there is also some interest in migration, the test sample may be sewn onto a colourless control material. The results are reported with respect to the specified test, describing the extent of colour change and staining of the control sample and expressed by reference to the Grey Scale [90, 100].
A pigment targeted for the spin dyeing or textile printing market must be fast to water [101] and seawater [102], irrespective of its method of application. Laundering tests are designed to evaluate the washfastness of textiles. Test cycles with standard detergents can last 30 min to 4 h at temperatures between 40 and 95 °C [103]. Laundering methods also include techniques to determine the colourfastness of a sample to peroxide containing washing agents such as sodium perborate [104], fastness to bleaching with hypochlorite [105] or to chlorinated water [106]. The fastness to perspiration is simulated through alkaline or acidic test solutions that contain histidine [107]. There are other tests concerning fastness to wet or dry rubbing [108], ironing [109], dry cleaning [110], solvents [111], acid [112] or alkali [113] and heating with sodium carbonate [114], including the fastness of a pigmented textile sample to bleaching with peroxide, hypochlorite or chlorite [115]. It is possible to simulate the conditions at each stage of the manufacturing process through a series of determinations on test solutions containing varying amounts of the active agent in combination with suitably adapted experimental parameters. Out of the large number of other standardized tests, the hot pressing test should be mentioned. It consists of a series of experiments, carried out for 30 s at 150, 180 and 210 °C under pressure. This test was designed specifically for synthetic fibres [116]. Occasionally, there is some interest in the vat fastness of a pigment that is used to spin dye fabrics such as viscose-rayon or cellulosics. This is usually determined in an alkaline dithionite solution containing ammonia at about 60 °C for 30 min. Samples are classified according to change of colour and the extent to which the colour is transferred onto other pieces of cloth.
1.6.3 Migration
The term migration refers to the occurrence of bleeding and blooming. Dissolved portions of pigment may migrate from their medium of application to the surface or into a similar material that their system is in contact with. Several seemingly related phenomena that may arise during the processing or application of a pigmented system that shows similar results, such as plate-out or chalking, are caused by different effects and are mentioned elsewhere (Sections 1.6.4.1 and 1.6.4.2).
The theoretical and application aspects of the mechanisms leading to migration have been studied particularly in plasticized PVC [117, 118]. From the results and mechanistic concepts, which are not limited to the materials and binders that were actually used in these studies, the following picture emerges: bleeding and/or blooming is only observed in a supersaturated solution of a pigment in its vehicle system. Several conditions promote blooming and bleeding:
- Processing of the pigmented system at higher temperatures, that is, creating a large difference between processing and application temperature.
- Partial or complete dissolution of the pigment in its medium (a polymer, a plasticizer or a mixture of these) at the processing temperature.
- The actually dissolved portion of pigment exceeds its solubility at the storage or application temperature. This condition occurs very frequently.
- Supersaturation after cooling, as mentioned above.
- Free mobility of the dissolved pigment particles in their medium at the temperature of application. This is a condition that frequently occurs if pigments are dispersed in plasticized PVC, polyolefins or other polymers that are applied at a temperature – usually room temperature – above their glass transition point. Polystyrene and polymethacrylate, on the other hand, whose glass transition temperatures are above 100 °C, do not migrate at ambient temperature. There are exceptions, however; typically stable pigments may start to migrate at the high concentrations and under the harsh experimental conditions under which tests are typically carried out (see, for example, Reference [119]). Migration is more likely to occur at high storage temperatures (120 or 80 °C). At ambient temperature, the rigid polymer matrix prevents dissolved pigment particles from moving, which makes it possible to even use soluble dyes without facing the problem of migration. The plasticizing effect of ‘pigment dissolution’ on polymers such as polyester may decrease the glass transition temperature and the crystallinity.
- Satisfactory crystal formation; that is, sufficiently high probability for new seed crystals to form. This implies that the already dissolved particles should not crystallize preferably or exclusively at the surface of the undissolved pigment particles.
There is some doubt about the frequently quoted migration or diffusion of solid pigment particles in plasticized PVC. The Kumis and Roteman equation [120], which describes the correlation between the free volume inside a polymer matrix and the diameter of diffusing substances, seems to preclude the possibility of migration of solid pigment particles. It is likely that the transportation of these solid particles is entirely unrelated to migration mechanisms based on diffusion phenomena. The effect probably arises in connection with a general streaming of plasticizer to the surface of the pigmented material, in which case it should be classified as a plate-out phenomenon [118].
Finally, the degree of migration is also controlled by the chemical constitution of the pigment (Section 1.4.4) and by its particle size distribution (Section 1.7.7).
1.6.3.1 Blooming
The term refers to the migration of dissolved pigment particles from the inside of a pigment/medium system to its surface, where they are deposited as a layer of pigment crystals. Even if it is rubbed away, this film will form again. The process is completed only after years, when most of the organic pigment has crystallized either on the surface of the pigmented material or inside it. Blooming is observed in various pigmented media whose application or processing involves high temperatures, such as baking enamels used in the coatings industry, metal deco printing for the printing industry, plasticized PVC, polyethylene, or rubber mixtures containing high-boiling naphthenic oils as plasticizers for the plastics market.
In a given medium and at constant temperature, the tendency of an organic pigment to bloom is a function of its concentration. Many systems have a concentration limit below or above which blooming does not occur. The range of concentrations at which blooming is observed widens with increasing processing temperature, which subsequently enhances the solubility of a pigment. This is indicated in Table 1.1, which shows the correlation between blooming, concentration and processing temperature for Pigment Red 170, a Naphthol AS pigment, in PVC containing 30% dioctyl phthalate (DOP) as a plasticizer. Table 1.2 gives specifications about the temperature dependence of pigment solubility.
Table 1.1 Blooming (+) of Pigment Red 170 in plasticized PVC in relation to the pigment concentration and the processing temperature (by F. Gläser).
| Processing temperature (°C) | Pigment concentration (wt%) | |||||
| 0.005 | 0.01 | 0.025 | 0.05 | 0.1 | 0.5 | |
| 140 | + | + | − | − | − | − |
| 160 | + | + | + | − | − | |
| 180 | + | + | + | + | − | |
| 200 | + | + | + | + | − | − |
Table 1.2 Solubility of Pigment Red 170 in plasticized PVC (by F. Gläser).
| Temperature (°C) | Solubility (wt%) |
| 20 | 1.8 × 10−7 |
| 50 | 1.2 × 10−5 |
| 100 | 4.0 × 10−4 |
| 120 | 1.3 × 10−3 |
| 140 | 3.7 × 10−3 |
| 160 | 9.7 × 10−3 |
| 180 | 2.3 × 10−2 |
| 200 | 5.1 × 10−2 |
Blooming is one of the features that differentiate pigments from dyes. Fluorescent dyes can only be applied in polymers such as plasticized PVC or polyolefins up to a certain concentration limit. Dyes dissolve in these polymers even at ambient temperature, preventing crystallization and thus precluding blooming. The solubility limit at room temperature can only be exceeded if dye concentrations are very high. In that case, blooming may occur.
Pigment concentration is not the only system-dependent migration determinant; the entire composition of the coloured medium plays a role. The tendency of plasticized PVC to promote blooming, for instance, depends on a series of parameters, such as the polymer itself, its synthesis including possible use of emulsifiers, the K value, choice of plasticizers and stabilizers, and their concentration. Specialized agents such as polymer plasticizers or intermediate layers designed to block particle movement can reduce or prevent migration.
Figures 1.27 and 1.28 show the mechanisms underlying the dissolution and crystallization processes during manufacture and storage. As above, the example chosen to illustrate this principle is Pigment Red 170 in plasticized PVC [117].

Figure 1.27 Transmission spectrum of P.R.170 in plasticized PVC; processing temperature: 180 °C.

Figure 1.28 Transmission spectrum of P.R.170 in plasticized PVC; processing temperature: 140 °C.
Transmission spectra were taken immediately after allowing the pigmented samples to cool to room temperature. Interpretation of the characteristic 570 nm band shows that at 180 °C all of the pigment crystals are dissolved, provided their concentration is small (0.005 and 0.01%, respectively). At 140 °C, however, not all crystals are dissolved. Figure 1.29 demonstrates that crystallization, taking place inside a sample rather than on its surface, is not completed even after years of storage [117].
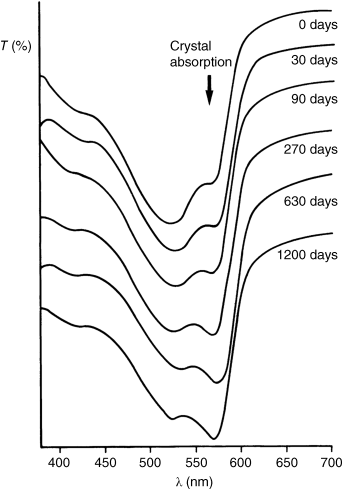
Figure 1.29 Transmission spectra of Pigment Red 170 in plasticized PVC in relation to the storage time (pigment concentration: 0.01%, processing temperature: 140 °C). The curves are shifted relative to each other in the direction of transmission.
Scanning electron microscopy makes it possible to trace the time curve of blooming on the surface of a plasticized PVC sample [121]. Pigment Yellow 1 develops detectable surface crystals within a period of only a few hours, and the area is densely covered within a day (Figures 1.30 and 1.31). Even a small space may be sufficient for a pigment to develop various apparently different crystalline forms (Figure 1.32), although only one crystal modification appears by X-ray diffraction analysis. The tendency to bloom renders a series of pigments unsuitable for use in such systems.

Figure 1.30 Scanning electron micrographs of a plasticized PVC surface with bloomed Pigment Yellow 1 after different storage times (a) and (b). Pigment concentration: 0.2%. (c) and (d) Magnified images of (a) and (b), respectively.

Figure 1.31 Scanning electron micrographs of a plasticized PVC surface with bloomed Pigment Yellow 1 after different storage times (a) and (b). Pigment concentration: 0.2%. (c) and (d) Magnified images of (a) and (b), respectively.

Figure 1.32 Scanning electron micrograph of a plasticized PVC surface with bloomed Pigment Red 3 after 84 days. Pigment concentration: 0.5%, processing temperature: 180 °C.
Basically, blooming and bleeding are thought to be a function of pigment constitution. Monohydrazone pigments of comparatively low molecular weight, such as P.Y.1, migrate easily. It is not possible to extrapolate the experimental results concerning one pigment to predict the behaviour of another, even if they are chemically related. This is especially true for concentration limits.
Pigments that bloom also bleed easily. The reverse is not always true: there are several pigments that are somewhat prone to bleeding but which do not bloom.
1.6.3.2 Bleeding/Overspraying Fastness
A pigment is said to bleed if some portion of it migrates from the application medium in which it is partly dissolved to a similar colourless or differently coloured medium with which it is in contact. This phenomenon is of particular concern to the plastics and coatings industry.
Unsurprisingly, therefore, the phenomenon of bleeding has given rise to various industrial standards, including generally accepted terminology [122, 123], standardized tests to determine the tendency of a material to migrate and the rate of plasticizer migration [124], evaluations of bleeding into coloured paper, cardboard and carton [125], plastics in general [126] and plasticized PVC in particular [119]. The samples used in each of the standardized tests must be prepared according to exact specifications. This is particularly important considering the fact that both the entire thermal treatment and the surface structure of the polymer may influence the tendency of a dissolved pigment to migrate.
The extent of bleeding in a plastic is evaluated with a test known as the sandwich method. The test sample is placed between a white, smooth surface of plasticized PVC on top and a standardized sheet of filter paper underneath. The resulting sandwich is then placed between layers of foam material, which in turn are covered by glass plates. The resulting sandwich is allowed to remain in an exposure chamber at 50 °C for 72 h [126].
Plasticized PVC is tested by covering both sides of the pigmented test sheet with white plasticized PVC films, which are placed between glass or aluminium plates. With a certain weight on it, this sandwich is exposed for 24 h at 80 °C (or for 15 h at 100 °C; or for 2h at 140 °C) [119]. The thickness of the test sample does not affect the test results.
The degree of bleeding is estimated either by colourimetrically determining the staining of the contact sheet and/or the filter paper [126], or visually by reference to the Grey Scale [100]. It is important to evaluate the results immediately after testing, because pigment diffusion into the contact sheet may make it impossible to maintain the colour during storage.
Tests to determine the bleed fastness of a paint system – also called overpainting or overcoating fastness – are not subject to industrial standards. A commonly used technique involves overpainting a white paint film of defined thickness to a full shade colouration of the test pigment dispersed in the same binder. Baking is carried out under defined conditions; heat-set paints, for instance, remain in a drying oven at 140 °C for 30 min. Pigment migration into the white material is determined visually or colourimetrically.
The commercial demand for pigments that tend to bleed is limited. A major consideration is the processing temperature in a particular medium. There are several baking paints, for instance, which do not bleed into a white coating if they are cured at 120 °C, while a certain extent of bleeding is observed at temperatures between 140 and 160 °C.
Likewise, in the paint and printing ink field, the properties of an entire system are equally important, including the composition of binders and the solvents. Amongst others, important variables are the rate of solvent release and the crosslinking of the binding agent, as well as the extent and duration of heat exposure. Pigments designed for use in such systems should be tested together with their vehicles and under conditions that relate to actual processing and use.
As mentioned above, increased pigment concentrations are associated with a tendency to bleed, especially in materials such as plastics, which makes it necessary to work below a certain limit value. Some media, such as plasticized PVC, can accommodate modest amounts of a bleed-sensitive pigment. Increasing amounts of plasticizer in plasticized PVC enhance the tendency of a pigment to bleed. Another concern is the choice of plasticizer. There are phosphoric acid triesters, for instance, which solubilize pigments somewhat more than compounds such as the frequently used dioctyl phthalate, which in turn exceeds the action of polymer plasticizers. The latter may even be used to reduce or prevent otherwise migrating pigments from bleeding. The use of a pigment with a certain tendency to bleed may present an economic advantage, if it is applied under conditions that preclude bleeding. There are also several applications that are not susceptible to bleeding; the pigmented polymer, for instance, may not come into contact with another suitable plastic material, or there may be no necessity to cover a pigmented paint film with a second (also pigmented) layer.
1.6.4 Disturbances during the Processing of Pigmented Systems
There are many ways in which an organic pigment may interact with its medium; both user and manufacturer frequently find that a pigment adversely affects the performance of an entire system. Polymer products are amongst the most sensitive in this respect. In pigment processing and application, where surface phenomena are important, the effect of pigment–vehicle interaction may be extremely aggravating and it is advisable to avoid concentrations and conditions that are prone to give rise to problems. Pigmented systems are particularly susceptible to the temperature, and knowledge of the mechanisms underlying pigment–vehicle effects might prevent an unwelcome surprise.
1.6.4.1 Plate-Out
The term refers to the deposition of pigment on the surface of the processing equipment or on the system itself. In contrast to similar phenomena caused by blooming, plate-out does not reappear once the pigment film is rubbed off.
Plate-out is commonly observed on the calendering and rolling equipment that is used to process pigmented PVC. It is caused by an incompatibility between components such as lubricants, stabilizers (especially those of a barium/cadmium basis), plasticizers or other materials by the PVC mixture. These materials, in migrating to the surface of the system, carry pigment particles along with them. Plate-out is thus a direct result of the incompatibility of certain additives with the PVC medium. As a consequence, rejected particles are deposited on the surface of the calender rolls or other processing equipment, covering it with a coloured film.
Conditions to be avoided are high temperatures and high pigment concentration, both of which increase plate-out effects.
The phenomenon appears to be particularly sensitive to the wettability of the pigment particles by the polymer. Types with higher specific surface areas are more subject to plate-out than varieties with a larger particle size [127, 128]. Likewise, plate-out phenomena are also observed in powder coatings, which frequently contain pigment particles or other solid particles that are not suitably wetted. Such coatings emerge from the curing process covered by a layer of pigment, which may also include crystals of the curing agent. Poor rheology must be expected to present problems with regard to the curing process by making it more difficult for the binder to wet the surfaces of the particles. Within seconds, the molecules begin to crosslink and the material is thus cured at a temperature at which it is expected only to melt and sinter. This phenomenon not only causes a rapid viscosity increase but also fails to allow sufficient time for the vehicle to completely wet all particles. Attempts to remedy this problem have resulted in the development of specialized hardeners that react only at elevated temperatures, considerably reducing the tendency of a material to exhibit plate-out.
1.6.4.2 Overpigmentation/Chalking
Surface degradation of a pigment–vehicle system results in an effect known as chalking, a familiar phenomenon in connection with the artificial or natural weathering of paint films that contain titanium dioxide. The chemical reactions and micromorphological processes underlying this effect have been studied extensively [129, 130]. In the presence of moisture, the photoactive titanium dioxide pigment, acting at the interface between pigment and binder, adds to the natural decomposition of the binder through daylight UV radiation. Increasing weathering time thus separates the pigment from its medium and degrades the surface of the pigment–vehicle system to such an extent that previously dispersed pigment particles come to the surface. Chalking is facilitated by using insufficiently processed pigment types or by working pigments into carrier materials that do not have good weatherfastness. The degree of chalking on a given surface can be determined by the Kämpf method [131] or the adhesives technique [132], both of which also lend themselves to the analysis of degraded paint films.
Similar phenomena are observed in enamels that contain certain organic pigments, but it should be noted that the chemical and micromorphological processes that are responsible for this form of molecular degradation are not known.
Despite the fact that several lightfastness and weatherability studies have reported that coatings made from chalking pigments absorb appreciably more heat than do comparative non-chalking systems, the exact role of the temperature in decomposing the vehicle is not completely clear. For paints containing organic pigments, the degree of chalking may be determined in analogy to TiO2-containing coatings.
In the plastics industry, exceeding the limit of pigment incorporation into its vehicle is likely to give rise to chalking phenomena in the broader sense of the word, particularly with organic pigments that exhibit a high specific surface area. In this case, the typically long-chain polymers or PVC plasticizers are unable to suitably coat and fix the pigment particles, a problem that has an added degree of complexity in the presence of additives or other pigments, such as TiO2 or extenders.
The effect of chalking is rapid and marked even after a short weathering time. There appears to be no clear distinction between chalking in the broader sense of the word and plate-out; the two phenomena are very much related. As in plate-out, and unlike in blooming, the particles that adhere to the polymer surface through chalking can be removed comparatively easily without reappearing.
1.6.4.3 Distortion/Nucleation in Polymers
Many organic pigments, like several other compounds, are able to induce nucleation in partially crystalline polymers, a phenomenon commonly observed in certain polyolefins. The surfaces of foreign particles such as pigments that are contained within a polymer material provide crystallization nuclei that initiate crystal growth. It is not only the crystallization rate, however, that is influenced by nucleating agents, but also the morphology and thus the mechanical properties of their host compound. Nucleating agents can even have a devastating effect on extrusion moulded plastics articles. This may be exemplified by the comparatively drastic consequences that it has for strong, thick, usually large, nonrotation-symmetrical extrusion parts, such as bottle crates. A distorted crate that will not fit onto any other is worthless. Rotation-symmetrical parts, on the other hand, may suffer from increased internal stress. The consequence of abnormal shrinkage can range from stress crack corrosion to long-term damage through weathering. The performance of a material in terms of tensile strength, breaking strain and impact resistance is more or less affected by nucleation [133, 134], which is as much a function of the time–temperature curve as it is of pigment concentration and of the choice of polymer. The shape of the pigment particles is another major factor; acicular particles are usually orientated along the direction of the polymer melt and give rise to uncommon nucleation effects. It appears that the surface structure of the pigment particles forces the adsorbed polymer molecules to grow into spherulites, as demonstrated in Figure 1.33.

Figure 1.33 Image of a crystallization nucleus triggered by a pigment particle in polyethylene in polarized light, taken with an optical microscope.
1.6.5 Dispersion
1.6.5.1 General Considerations
Organic pigment powders are normally sold as agglomerates (Section 1.5), ready to be incorporated into the medium of application. This process, known as pigment dispersion, refers to the distribution of a pigment throughout the application material, accompanied by a reduction of the agglomerate size to afford primary particles and aggregates, or at least smaller agglomerates. A pigment is referred to as being more or less easily dispersible, depending on the mechanical forces that have to be applied to achieve optimum dispersion. Reducing the size of agglomerates is necessary to develop the best possible application properties in a given pigment or pigment–vehicle system, whose performance is largely controlled by the particle size distribution. The effect of dispersion on the behaviour of a pigment vehicle system is comparable to the effect of simply reducing the average pigment particle size. The pigmented system will:
- exhibit increased tinctorial strength, particularly in white reductions;
- undergo a change of shade;
- be more transparent/less opaque;
- provide enhanced gloss;
- increase in viscosity;
- and, finally, feature a reduced critical pigment volume concentration, which is important in mill bases.
A certain amount of shearing forces have to be applied to overcome the surface forces that maintain the adhesion between agglomerated pigment crystals. In practice, the shearing forces that are necessary to reduce the particles in a given pigment sample to smaller or even optimal particle size, that is, the dispersibility of a pigment powder, depends on several factors:
- Pigment-related aspects, which involve the chemical constitution, crystalline modification, particle size distribution, particle shape, surface structure, preparation and processing of the pigment powder, especially in terms of drying and milling.
- The chemistry and physics of the vehicle and its components, including factors such as polarity, molecular weight or molecular weight distribution and viscosity. The dispersibility of a system that contains several different components such as resins depends on the solubility of each individual component in the medium and their compatibility with each other.
- The chemical or physical characteristics of solid or liquid additives, such as extenders, white pigments, dispersing agents, plasticizers or pigment/extender combinations.
- Interactions at the pigment–vehicle interface.
- Design of the dispersing agent and its mode of operation in combination with the dispersion conditions.
- Pretreatment of the pigment prior to the dispersion process, including time, temperature, wetting and other parameters connected with incorporation of the pigment in the vehicle.
- Formulation of the mill base, including the pigment volume concentration (PVC).
The dispersion process may be viewed as simultaneously pursuing four different objectives:
- Agglomerates are broken down through mechanical shearing forces. Particle size reduction: desagglomeration.
- The surface of the pigment particles is wetted by the binder and by other components of the medium: wetting.
- The resulting wetted pigment particles are distributed throughout the entire medium: distribution.
- The pigmented system, which consists of dispersed particles, is stabilized to preclude reagglomeration and/or flocculation: stabilization.
These four facets of the dispersion process are discussed below.
1.6.5.2 Desagglomeration of Pigment Particles
According to Rumpf [135], there are four ways in which agglomerates can be broken down:
- between two solid surfaces (dry milling in roll mills or roll crushers);
- by impingement, that is, by imparting kinetic energy to a material and crushing it through impact on a solid surface (dry milling in impact mills);
- by mechanical energy that is transferred onto the material through the surrounding medium (wet grinding);
- by thermal crushing.
Pigments that are incorporated in a medium are practically always crushed by mechanical means, that is, the agglomerates are broken down through shearing forces. The pigment particles are thus broken down through their medium and not through direct impact between each other.
Sometimes the dispersion process may inadvertently result in destruction of the aggregates and the primary particles, which happens particularly with pigment powders consisting of coarse or acicular particles – not forgetting the effect of intensive shear [136].
1.6.5.3 Wetting of Pigment Particle Surfaces
There are two aspects to the wetting of pigment particles:
- the spreading of a liquid or a binder system over the surface of the pigment particles;
- the soaking of the pigment powder, especially of the agglomerates, with liquid components.
The wetting operation is primarily a function of [137]:
- the energetics of the interaction between the pigment surface and the medium;
- kinetic parameters (such as diffusion and wetting rate);
- the steric sizes and geometry of the components, including the porosity of the agglomerates and the molecular size of the wetting agents;
- the rheology of the wetting medium.
The interactions between the surface of the pigment particles and their surrounding medium are of prime consideration in the wetting process and are optimized in the development and production of easily dispersible pigments.
The energetics of the wetting of particles have been discussed extensively [138–140]. The specific thermodynamics of interfaces have been studied for several organic pigments and other materials [141–143]. This approach provides information as to whether a liquid will spread over the surface of a solid on its own, whether the operation requires energy or whether a surface is not wettable at all. It is possible to find the surface tension of a pigment (γ1) by measuring and comparing contact angles (α), which reflect the relative attraction of the liquid for the solid compared to the attraction of the liquid for itself. The experimentally determined surface tension of the liquid (γ2) is the basis from which to calculate the interfacial tension (γ12) and the wetting tension (β12 = γ2 cos α) by using the following equation:
Thus, the greater the value of β12, the easier it is to wet the surface. The kinetic aspect, that is, the actual wetting rate or wetting time, is quantified by the modified Washburn equation, a simplified version of the Hagen–Poiseuille equation for cylindrical capillaries in the case of incomplete wetting [144, 145]. In using this equation, wetting is always assumed to be thermodynamically allowed [142]:
in which:
| t | = | penetration time |
| α | = | contact angle |
| γ2 | = | surface tension of the liquid |
| γ2 cos α | = | wetting tension |
| l | = | capillary length |
| η | = | viscosity |
| r | = | radius of the pores within the agglomerates |
| V | = | transported volume of liquid |
According to the above equation, the time t required to wet a powder is proportional to the viscosity of the surrounding medium, the depth of the pores and the transported volume. The wetting time, on the other hand, is inversely proportional to the wetting tension – the driving force that promotes wetting – and to the cube of the radius of the pores in the agglomerates.
The viscosity of the binder is thus the only variable wetting time determinant during pigment processing: both r and l are pigment-specific structural parameters and are therefore constant, which is also true for the wetting tension. The viscosity, on the other hand, can be changed systematically by modifying the composition of the mill base or by increasing the dispersion temperature.
The efficiency of any dispersion operation is essentially a dual function of the breakdown of the agglomerates and the wetting of the pigment surface. One or the other of these two barriers will usually dominate, depending on the medium and the dispersion equipment. Dispersing a pigment in a polymer such as PVC or a polyolefin, for instance, is largely a question of breakdown of the agglomerates through mechanical action.
To demonstrate this tendency, samples of the gamma form of Pigment Violet 19 (Section 3.2.4) were dispersed on a roll mill into PVC containing varying amounts of plasticizer (Figure 1.34) [146].

Figure 1.34 Dispersion of Pigment Violet 19, γ-modification, in PVC on a roll mill in relation to the plasticizer content (DOP) and the dispersion temperature. PVC: K value 70.
The tinctorial strength of the pigment–vehicle system is plotted against the dispersion temperature. The tinctorial strength is an indication of the quality of pigment dispersion in the end product. The resulting graphs illustrate the correlation between pigment dispersion, temperature and plasticizer content. Increasing the amount of plasticizer within a polymer appears to reduce the tinctorial strength, reflecting poorer dispersion. The second diagram, showing the tinctorial strength that is attained through a certain plasticizer content at the lowest possible temperature (130 °C), demonstrates the decreasing influence of heating as more plasticizer is added. Obviously, the quinacridone pigment being used in this study does not disperse easily in its medium. The result is somewhat surprising, since this particular plasticizer is known to wet organic pigments easily. One might therefore expect an increased dioctyl phthalate concentration to increase rather than decrease the dispersibility of the pigment. It can thus be concluded that in this case the amount of shear being imposed on the system appears to be the primary determinant. If the plasticizer content in a polymer is increased, its rheology will change, which is associated with a stronger plastification of the polymer and consequently reduced shearing forces. This is in aggreement with several rheological studies on a series of these PVC–plasticizer mixtures, in which additional shear was applied through temperature changes (Figure 1.35).

Figure 1.35 Dispersion of Pigment Violet 19, γ-modification, in PVC at varying plasticizer content. Relation between the shearing forces in the plasticized PVC during pigment dispersion and the degree of pigment dispersion (depth of shade) after 8 min of dispersion.
Wetting out a pigment for several days by simply storing the manually prepared pigmented PVC paste (DOP content: 39%) makes for almost optimum dispersion, which requires very little shear (see Figure 1.92, Section 1.8.3). The wetting of the surface of the pigment particles by the plasticizer molecules thus determines the outcome of the dispersion process.
For all practical purposes, shear continues to be of prime consideration in pigment dispersion, even if the wetting tension of the medium is high. Therefore, unsurprisingly, the degree of agglomeration in a dispersion product varies according to the design of the equipment with which it is produced. Most of the dispersion equipment that is used in industry today has received appreciable theoretical and experimental consideration regarding both machinery and operation and has been evaluated on various pigment–vehicle systems [147–149].
Increasing the temperature frequently leads to better wetting, while the reverse is true for the mechanical breakdown of agglomerates. Similar observations have been made regarding the effect of wetting and dispersion agents [150].
To illustrate this trend, a sample of the β-modification of Pigment Violet 19 was dispersed in an offset vehicle on a three-roll mill. Two series of determinations, one at constant temperature and one at constant shearing force, afforded data on the dispersibility of the pigment powder in relation to the shearing force (applied in the nip between primary and secondary rolls) and to the dispersion temperature. The resulting printing inks were more or less thixotropic; their viscosities were measured at 23 °C [147] after the thixotropic effect had levelled off (Section 1.6.8.1). Figures 1.36 and 1.37 plot viscosity values derived from flow curves that were taken at a constant shear rate of D = 220 s−1 versus the pressure between the rolls and the temperature, respectively.

Figure 1.36 Effect of the pressure between the rolls during isothermic dispersion (23 °C) of 20% Pigment Violet 19, β-modification, in a sheet offset vehicle (six passes) on the viscosity of the printing ink.

Figure 1.37 Effect of the roll temperature and the number of passes during the dispersion of 20% P.V.19, β-modification, in a sheet offset vehicle at constant pressure between the rolls on the viscosity of the printing ink.
Evidently, the effect of the temperature increase during dispersion is more significant than the shearing stress or shearing force created by the rolls. The plots demonstrate that the degree of dispersion afforded by even a slight temperature increase exceeds the effect of several additional passes on three-roll mills, even at maximum shearing force. However, the shape of the graph also demonstrates that the degree of dispersion is still far from optimum: the tinctorial strength of prints made from these inks continues to increase considerably under comparable conditions.
Average temperatures (40–70 °C) in combination with high-shear dispersion equipment, of which three-roll mills are a good example, appear to afford the best results in terms of pigment dispersion in wetting systems, such as offset varnish. Figure 1.38 shows a curve in which the degree of dispersion reaches a distinct maximum at one particular temperature.

Figure 1.38 Effect of dispersion temperature (roll temperature) and pressure between the rolls during dispersing of Pigment Red 53:1 in a sheet offset varnish (pigment concentration: 20%) in a three-roll mill on the degree of pigment dispersion. This correlation is illustrated by showing the viscosity of the printing inks at a shearing rate D = 220 s−1.
1.6.5.4 Distribution of the Dispersed Pigment in its Medium
Achieving a statistically uniform distribution of pigment particles throughout all volume units of the entire application medium is a question of the equipment and its mode of operation and also of the conditions under which the dispersion process is carried out.
1.6.5.5 Stabilization
One of the primary concerns in the present discussion is the necessity to stabilize the system of wetted pigment particles in their vehicle system so that the dispersion process does not reverse itself. Stabilization, that is protection against reagglomeration and/or flocculation, is a somewhat complex matter (Section 1.7.5) [151] and depends on various parameters, including the composition of the medium, especially the choice and amount of solvents, the chemical and physical features of the different components, the viscosity of the vehicle system and the effect of adsorptive layers covering the surface of the pigment particles. The complexities of attraction and repulsion are discussed at great length in the literature. Three of these concepts should be mentioned. All of them regard adsorption as a key process in stabilization, but differentiate between ionic and steric forces:
- The DLVO theory, a quantitative theory of colloid fastness based on electrostatic forces, was developed simultaneously by Deryaguin and Landau [152] and Verwey and Overbeek [153]. These authors view the adsorptive layer as a charge carrier, caused by adsorption of ions, which establishes the same charge on all particles. The resulting Coulombic repulsion between these equally charged particles thus stabilizes the dispersion. This theory lends itself somewhat less to nonaqueous systems.
- A mechanism claiming steric interaction between the molecules adsorbed at the solid–liquid interface [154] was first proposed by Crowl [155].
- The entropic repulsion theory of Clayfield and Lumb [156] suggests that the bulk of the protective macromolecular layer prevents other particles from approaching close enough for the attractive forces to cause agglomeration [157].
1.6.5.6 Dispersion and the Critical Pigment Volume Concentration
The term pigment volume concentration (PVC) refers to the percentage of the volume of a pigment that constitutes the nonvolatile portion in a formulation. The critical pigment volume concentration (CPVC), on the other hand, indicates the proportion of the volume of a pigment to the volume of the vehicle at which there is just sufficient binder to wet all pigment particles and to fill the voids between them, leaving no excess vehicle [158, 159]. The CPVC has received considerable attention in connection with operations such as optimizing the formulation of mill pastes, a process in which cost–benefit considerations are a primary concern [160]. The CPVC decreases with increasing degree of dispersion and hence with increasing surface area that is available for wetting by the binder.
This principle has been illustrated in a study designed to show that the CPVC is affected by changes in the dispersion conditions (Figure 1.39) [161].

Figure 1.39 Effect of the dispersion temperature and the degree of dispersion of P.Y.13 on the viscosity and CPVC of sheet offset inks. Viscosity measurement at 23 °C.
A sample of Pigment Yellow 13 was dispersed in an offset vehicle. A series of printing inks, each with a different pigment concentration, were produced on a three-roll mill in four passes at temperatures of 15, 30 and 45 °C, respectively. Flow curves were taken after 24 h at 23 °C; the respective viscosities for D = 220 s−1 had to be determined indirectly by interpolation. The CPVC was then determined by plotting viscosities against pigment volume concentration and finding the point at which the viscosity values level off [161]. By definition, this is the point at which all binder molecules are adsorbed at the pigment surface and are thus unavailable to wet additional pigment surfaces: the system is now immobile.
Several parameters change abruptly at the CPVC. Experimental data on the influence of binders, additives and suitable pigment preparation are shown in Figure 1.40, which is a plot of the viscosity versus pigment volume concentration curves for three samples of one pigment, incorporated in three different offset varnishes [162].

Figure 1.40 Influence of the binder on the CPVC and of the binder and the CPVC on the viscosity of P.Y.13 in sheet offset vehicles of different composition at constant dispersion conditions in a three-roll mill.
Dispersed pigment formulations must be adequately prepared to afford products with optimum commercial properties. This concerns both the application of concentrates and the conversion of concentrates into end-use products, which should preferably exhibit good flow properties in combination with high tinctorial strength. Knowledge of the above correlations and mechanisms aid the manufacturer in achieving optimum product performance.
1.6.5.7 Test Methods
Manufacturers of organic pigments frequently use additives in an attempt to improve parameters such as the wetting time and wetting volume and thus to enhance the dispersibility of their products. The decision about each individual approach is usually made on the basis of empirical data concerning the application properties of the products. This is understandable in view of the fact that the dispersibility of a pigment is known to depend on its medium and on the conditions under which it is applied, a problem that is so complex that it is solved best by carrying out a series of pilot-scale experiments in different vehicles.
Where pigment dispersibility is to be quantitatively related to other parameters, it is often useful to indirectly determine the degree of dispersion and the particle size. The technique of measuring the quality of dispersion that is achieved at certain marked points in the procedure – which may be defined in terms of time, passes or cycles – is a feature common to all of these methods. Alternatively, it is also possible to relate the dispersibility of a material to a series of different processing temperatures, which is of particular interest to the plastics industry. The list of possible criteria includes tinctorial strength, gloss, transparency, viscosity and other application properties of the pigment–vehicle system.
A different principle is pursued by methods that rely on direct particle size determination by optical microscopes or other equipment. However, it is not always possible for direct methods to recognize all of the particles in organic pigments, because they can be much smaller than 1 µm. Beyond a certain degree of dispersion, the only information that can be extracted by direct methods relates to the still existing coarser portion of the pigment agglomerates, which is not normally recognized when application properties are determined. Additional information may be obtained by similarly measuring the particle size in a paint or an ink by examining the respective pigmented application media with grindometers [163, 164]. To control the production and to test new formulations, grindometers are useful as long as the particle size is larger than 1–2 µm. No useful information can be given for pigments with a degree of dispersion smaller than 1 µm, but this range provides the primary determinants of tinctorial strength, gloss and other aspects of pigment application properties, which restricts the use of grindometers. They are, however, used to advantage in predicting possible processing problems caused by the pigment and are employed, for instance, by producers of offset printing inks to analyse layers around 1 µm (Section 1.8.1).
The manufacturer who wishes to characterize the dispersibility of a given organic pigment batch frequently refers to curves plotting the tinctorial strength versus dispersion time. Industrial standards [165] have been developed for several dispersion processes and instruments, their primary operational parameters, and data evaluation. Other standard tests provide information regarding individual applications: specific tests have been designed for pigment dispersion in low-viscosity alkyd resin systems that are dried by oxidation, in heat-set alkyd/melamine resin systems and also in highly viscous binders dried by oxidation [166]. The mill base is sampled at certain points during the dispersion process, which are either referred to simply by time or defined through equipment-related units such as the repeated passage of material through the mill. The resulting samples are combined with a white pigment or a white pigment paste and the white reductions evaluated photometrically. The tinctorial strength presumably reflects the degree of dispersion. Although this is a seemingly straightforward method, the results may be deceptive if particles flocculate as white pigment is added or if the dispersion process is reversed, distorting the measurements. Such problems are readily revealed by the rub-out test, which determines the difference in tinctorial strength between a rubbed and a nonrubbed area. A deviation of more than 10% suggests that the determination be carried out at the rubbed area.
The tinctorial strength of a pigment is not normally proportional to the dispersion time, but generally increases hyperbolically [167]. Since all pigment batches are expected to contain a more or less large portion of agglomerates, held together by weak forces, it is not surprising that the initial dispersion rate is comparatively high. The agglomerates are, thus, the first units to be broken down, leaving a material composed of less easily dispersible parts. This explains why the dispersion rate slows down and the dispersion–time curve asymtotically approaches an upper limit (Figure 1.41).

Figure 1.41 Dispersion of Pigment Violet 19, β-modification, in an alkyd-melamine resin baking enamel with a paint shaker. Reduction 1 : 2 with TiO2. Tinctorial strength–time curve.
This limit, which might be referred to as the ‘ultimate tinctorial strength’, reflects the maximum degree of dispersion that can be achieved in a particular vehicle system under a certain set of conditions. However, experimental results may deviate more or less from the theoretical concepts; an ideal dispersion is not normally realized: not all agglomerates are broken down entirely. This, however, is of no consequence, because even the experimentally determined ultimate tinctorial strength is by no means considered a standard for industrial application: technical operations are not always allowed to go to completion, and the dispersion process is often discontinued, mainly for economic reasons.
Although several concepts can be illustrated conveniently by plotting the tinctorial strength versus time, the resulting curves do not necessarily reflect a quantitative relationship. Not only is it impossible to accurately predict the ultimate tinctorial strength, but the curve also fails to reflect the shearing forces that are necessary to afford a certain degree of dispersion. Moreover, as dispersion time increases, the shape of the curve is often increasingly controlled by side effects.
A graph more suitable for quantitative evaluation is easily drawn up by transforming the above correlation into a linear one. The inverse of the tinctorial strength is simply plotted versus the inverse of the degree of dispersion, referred to in terms of time or some other unit [168–170]. The resulting doubly reciprocal presentation can be extrapolated to the intercept with the ordinate. This is the point at which the dispersion time is infinite and its inverse is 0, so that the height of the intercept equals the inverse of the ultimate tinctorial strength. A low gradient is associated with easily dispersible pigments, while a steep slope reflects increased shear requirements.
A characteristic number may be derived from the difference between two points, reflecting the increase in tinctorial strength, for example between two dispersion periods.
This characteristic, known as the dispersion hardness (DH), indicates the effort that is necessary to disperse a pigment [171]:
in which F1 is the K/S value measured at an early point on the curve, preferably chosen such that the degree of dispersion just allows the pigment–vehicle system to be completely homogeneous. F2 represents the K/S value at the second degree of dispersion, at which at least 90% of the ultimate tinctorial strength (F∞) is reached.
The tinctorial strength in white reductions is thus quantitatively defined by the Kubelka–Munk relation between the spectral absorption coefficient (K) and the spectral scattering coefficient (S), in which β∞ refers to the reflection of a completely opaque layer. The ratio K/S is proportional to the tinctorial strength:
The dispersibility of a pigment in a particular vehicle system is also reflected by the viscosity of the pigmented medium and by the gloss that it produces in application. Viscosity and gloss are frequently considered more suitable parameters to indicate the state of dispersion of a pigment in an application medium than the tinctorial strength, which reaches its optimum at a point at which other parameters, such as gloss, can be improved.
Standardized tests have been designed to determine the development of gloss in a pigment–vehicle system with time [172]. The reflectance values [173] of a coating or a print are plotted against the dispersion energy, measured in units of dispersion time or some other parameter. Since the resulting curve is as unsuitable for quantitative evaluation as the one derived from tinctorial strength determinations, plotting the inverse values is the approach that first comes to mind – and it is similarly useful in this case. The resulting linear correlation can be interpolated graphically to afford information regarding the dispersion energy that is necessary to produce a certain reflectance value, or to indicate the reflectance value that is realized at a particular point in the dispersion process.
Other effects, such as pigment recrystallization, also frequently influence the different partial processes of dispersion. Recrystallization may be defined in this context as a growth of larger particles at the expense of the smaller ones. Recrystallization is commonly associated with colouristic changes; it particularly improves the opacity and accordingly reduces the transparency of a material. Figure 1.42 outlines a tinctorial strength–time curve that is affected by recrystallization. In this particular experiment, Pigment Orange 43, a naphthalenetetracarboxic acid pigment (Section 3.4.2.4), was dispersed in an oscillating shaking machine (paint shaker) at 20, 40 and 60 °C, respectively, in a toluene-based publication gravure printing ink. Printed samples (cell depth = 18 µm) made from these inks were evaluated colourimetrically. As was expected, longer dispersion times evidently increase the particle size through recrystallization, decrease the tinctorial strength and cause a shift to more yellowish shades.

Figure 1.42 Dispersion of 5% Pigment Orange 43 in a toluene-based gravure printing vehicle with a paint shaker. Effect of the dispersion time and the dispersion temperature on the depth of shade (a) and on the hue of the prints (b).
Moreover, the crystal modification of a polymorphous pigment may change during the dispersion process. This was exemplified by incorporating a sample of the epsilon modification of copper phthalocyanine blue into a thermosetting coating system. The mill base contained pigment, alkyd resin, xylene (56%) and aliphatic hydrocarbons (10%); a paint shaker was used to disperse the pigment in the medium. Thermostats maintained a nearly constant temperature so that the difference between the readings for the mill base and the thermostated bath immediately after the dispersion process did not exceed 2 °C. The increase in tinctorial strength with time was determined colourimetrically after the coatings had been reduced 1 : 50 with TiO2, applied and dried (Figure 1.43).

Figure 1.43 Dispersion of Pigment Blue 15:6 in an alkyd-melamine resin baking enamel. Effect of the dispersion time and the dispersion temperature on the colouristic properties (depth of shade). White reduction 1 : 50 TiO2.
Likewise, Figure 1.44 illustrates the corresponding change in shade with increasing dispersion time.

Figure 1.44 Dispersion of Pigment Blue 15:6 in an alkyd-melamine resin baking enamel. Effect of the dispersion time and the dispersion temperature on the colouristic properties (hue). White reduction 1 : 50 TiO2.
The complex mechanism underlying this dispersion process of the epsilon modification of phthalocyanine blue is conceptually resolved into three steps:
- The dispersion itself, which consists of breaking down the agglomerates and covering the pigment surface with the binder components: a fast step in this example. The tinctorial strength thus increases rapidly. As the dispersion process proceeds, other mechanisms gain importance, such as the next two.
- A change in the crystal modification from the very reddish blue epsilon form to the greenish blue beta modification. The hue shifts accordingly, which is possibly also connected with a change in tinctorial strength.
- Recrystallization of the pigment particles: this is also associated with a decrease in tinctorial strength and a shift from a very greenish blue to a reddish blue shade.
1.6.5.8 Flush Pastes
Flushing is sometimes considered an alternative to the dispersion process, because it is the direct transfer of pigments in an aqueous phase, as they emerge from the synthesis, to a nonaqueous phase without previously drying and milling the colourant. The nonaqueous phase commonly consists of binders, such as alkyd resins, mineral oils, celluloseacetobutyrate or other suitable water-insoluble vehicles. The idea is to displace the adsorbed water from the surface of the pigment particles and to replace it by the vehicle components.
There are certain advantages to flushing, especially since it avoids drying the pigment presscake and milling the dry pigment, steps that are known to promote reagglomeration. The technique has been extensively investigated [174].
Alkali Blues (Section 4.1.1), which are highly polar, are particularly prone to reagglomerate and are difficult to disperse in dried form. Therefore, printing ink manufacturers prefer to use them as flush pastes at a pigment concentration of around 40%. Flush pastes of other organic pigments, such as diarylide yellow pigments for letterpress and offset printing, are less common in Europe. They are used for these purposes only in the USA and to a lesser extent in Japan, as well as by European branches of US companies. Since flushing generally produces dispersions of greater gloss and transparency than formulations produced with dry methods, products targeted for the letterpress and offset market were used also in Europe in flushed form before it became possible to manufacture highly transparent and glossy organic pigment powders for such printing inks.
Presscakes used for the flushing process typically contain between 15 and 20 wt% pigment. It is now also possible to purchase highly concentrated presscakes, containing 50 wt% and more. Despite the fact that they are recommended for use in aqueous media throughout the printing inks and coatings industry [175], such products only enjoy minor commercial recognition. It is difficult to colouristically standardize presscakes and to safeguard them against bacteria and fungi.
1.6.5.9 Pigment Preparations
The pigment industry today provides the user with a wide variety of pigment preparations to suit all purposes. In these pigment preparations, the pigment is in an already dispersed form. Purchasing a pigment in such a form considerably facilitates its application. Details regarding the composition, synthesis and application of such preparations are described under the respective applications (Sections 1.8.1–1.8.3).
1.6.6 Lightfastness and Weatherfastness
1.6.6.1 Definition and General Information
The lightfastness of a material is defined by the inherent ability of a given pigment–vehicle system to retain its initial colour value upon exposure to daylight. It is thus a system-related parameter that cannot be determined in connection with pure pigment only.
Although a few inorganic pigments are stable enough to withstand light completely and almost indefinitely, most inorganic and all organic pigments change their shade more or less readily upon illumination. The sensitivity of a given pigment–vehicle system is a function of pigment-related factors such as chemical constitution (Section 1.4.3), concentration, physical parameters (particle size distribution, Section 1.7.4, and crystal modification Section 1.5.3) on the one hand, and, last but not least, the chemical nature of the surrounding vehicle on the other hand.
Light is not the only power that may destroy or alter a pigment within its vehicle system. Even traces of water, gases or industrial effluents in the atmosphere may adversely affect the application properties of a material. The combined effects of weather and light will generally destroy a pigment faster than light alone. Weatherfastness has thus been defined in connection with the ability of a pigment vehicle system to withstand the chemical and physical factors inherent in weather, which also includes light. The exposure may be alternating or simultaneous. It is frequently recommendable and more common to refer comprehensively to the weatherfastness of a pigmented system instead of focusing on the effect of light alone.
Weatherfastness is defined as the simultaneous or alternating effect of irradiation and atmospheric impacts. Weather is difficult to express quantitatively, since it is a complex phenomenon and the response of a pigment–vehicle system to outdoors exposure depends largely on environmental conditions, such as the intensity of the sun, temperature, humidity, precipitation, oxygen concentration and the gaseous composition of the air. To complicate matters, all of these parameters are in turn functions of the time of day, the season and the location at which the determinations are carried out, including latitude, longitude, altitude, proximity of industrial plants and so on. It is evidently difficult to compare two or more sets of data, unless they are taken simultaneously in the same location, and there are no universally accepted weatherfastness standards. Both the lightfastness and the weatherfastness of a pigment–vehicle system are thus considered relative indications of pigment behaviour in a very strictly defined environment compared to a standard.
1.6.6.2 Evaluation Techniques and Equipment
Several lightfastness and weatherability test methods have in several countries been developed into industrial standards.
The Blue Scale, also known as the Woolen Scale, which was originally developed for the evaluation of exposed textile prints, serves as the standard of comparison mainly for graphic prints [176, 177]. It consists of eight blue-coloured woollen samples that differ in their fastness to light. The standards are prepared with chemically defined dyes whose chemical constitution is listed in the Colour Index. The swatches are graded according to numbers between 1 and 8, with 1 indicating extreme sensitivity to light, while highly resistant samples qualify for number 8. If lightfastness is to be evaluated in terms of a colour-change comparison, a defined standard and a sample are exposed together until the standard shows a significant change in colour, irrespective of whether it is the shade, the brightness and/or the intensity of the colour that is affected. A change equal to step 3 on the Grey Scale for Evaluating Colour Change is considered significant [90]. Several swatches may be tested simultaneously by exposing all samples to the same light source until one of the standards on the Blue Scale, for example the number 3, shows a noticeable change in colour. After partly covering the swatches (about ¼), exposure is continued until the next higher standard fades appreciably. The lightfastness test may be continued over the range of the Blue Scale up to step 6; the time is doubled in going from one standard to the one next in line.
The comparison with the Blue Scale may lead to different results, depending on how the standard and/or the pigment reacts to light. Blue dyes, for instance, are not at all uniform in their performance towards UV radiation [178]. This effect has been studied by using UV filters. The results point to the fact that although samples do not absorb differently over the visible range of the spectrum, steps 3–5 on the Blue Scale are considerably more sensitive to wavelengths under 335 and 360 nm than steps 1 and 2. Fading is therefore largely a function of the power distribution of the illuminant; an ageing light source or ageing filters may inadvertently influence the UV radiation, which may distort the results of an otherwise standardized test. Accelerated exposure devices thus have to be carefully controlled (see below), and location and even date and time should be recorded meticulously. Extremely lightfast materials, which correspond to steps 7 to 8 on the Blue Scale, present some unusual problems with regard to colour assessment. Differences between these samples are frequently subtle, difficult to determine accurately and rarely reproducible. The problem is aggravated by the fact that the textile carriers may not always be sufficiently stable.
Attempts have been made to replace the Blue or Woolen Scale by physical devices that measure the radiation of the light source. To determine the radiation of a given light source, the irradiance (in W m−2) is multiplied by the exposure time (in s). The equipment necessary to carry out such measurements is commercially available. Measurement in the UV region would be desirable. It has recently become possible to purchase an instrument that can measure spectrum-related irradiance in the important UV range.
Very light and weather resistant systems require very long outdoor exposure times. To accelerate the testing of pigmented systems, test methods have been developed that simulate outdoor conditions. The evaluation of such a test is not only conveniently accelerated, but the resulting data are easy to reproduce and independent of location, climate, date and time.
Thus, the compulsory requirement for automotive coatings of a two-year Florida exposure can be reduced to a period of less than 6 months. This is advantageous for the developmental process.
Today, xenon arc lamps are the most commonly used light sources for such tests. Xenon tubes are equipped with combinations of filters, which make it possible to approximate the radiation of natural sunlight. The filters can be combined as desired to alter the spectral distribution of the radiation, which may influence the outcome of degradation processes such as chalking. Figure 1.45 shows the energy distribution curve provided by a filtered xenon arc light compared to natural daylight. The filtered radiation emitted at high intensities by a xenon tube provides a good approximation to the energy distribution of normal daylight. Certain filter combinations screen out portions of UV radiation at specific wavelengths and thus approach daylight even more closely.

Figure 1.45 Energy distribution of solar radiation (according to CIE, No. 20) and filtered xenon arc light (Xenotest 1200) [179].
An industrial standard method has been developed to test the lightfastness of polymers in accelerated test equipment [180]. The apparatus consists of a quartz xenon tube with a special optical filter between the light source and the specimen to produce light that resembles window glass-filtered daylight [181]. Samples are mounted at a specific distance from the arc and are supported on a frame that revolves around the arc 1–5 times per minute for uniform exposure. A blower unit in the base provides a flow of air, which makes it possible to maintain a black panel temperature of 45 °C, measured by a black panel thermometer which is positioned at level with the samples. A black panel unit consists of a bimetallic thermometer mounted on a steel frame. Both faces of the frame plate and also the stem of the thermometer are coated with a heat-resistant glossy black enamel. The relative humidity level in the exposure cabinet is closely controlled.
Lightfastness is evaluated colourimetrically by comparing exposed coloured samples or portions of coloured samples with the original (unexposed) sample [182]. The colour difference between the two test specimens is expressed in CIELAB units [82, 83]. Instead of referring to the Blue Scale, results are described in terms of the radiation energy that is necessary to achieve a certain colour difference. Although ample data are available on the lightfastness of a large number of organic pigments in various media, the information usually relates to the Blue Scale. For the sake of uniformity and comparison, it therefore appears more appropriate to refer to the Blue Scale as a classification and evaluation system to describe specific pigments elsewhere in this book.
Accelerated exposure equipment may also be used to test for weatherfastness in plastic materials [183]. The natural destructive agents inherent in weather are approximated by filtering the radiation emitted by the xenon arc lamp and by spraying the sample with water under standardized conditions [183]. Test programs are designed to relate to actual outdoor exposure to rain and humidity. In a standard program, a 3 min wet cycle typically alternates with a 17 min dry period. Weatherfastness tests are carried out and evaluated like lightfastness tests: the black panel temperature and other parameters are the same in both procedures.
Weatherfastness tests on coatings have shown that prolonging the dry and wet periods, that is extending the usual 17 min dry cycle to a full 102 min and the wet period to 18 min, affords results that correlate much better with the Florida outdoor exposure tests. The humidity that penetrates a layer is known to interact with the various components in the coating, adversely affecting the mechanical properties of the material. The above-mentioned cycle apparently corresponds much better with the climatic conditions in areas like Florida. If recirculated water is used in accelerated exposure equipment, suspended particles may adhere to the surface of the coating, producing a thin film. This obviously has a detrimental effect on the test results.
It might be interesting to note how far the results of accelerated test methods agree with full-length outdoor exposure and the effect of natural daylight. The correlation between outdoor or daylight exposure, including factors such as location, elevation, proximity of industrial plants, maritime climate and so on, on the one hand, and accelerated methods, on the other hand, have been studied extensively on a wide variety of pigment–vehicle systems.
The results show that although the xenon arc radiation is adjusted by filters, there is still some problem in comparing colouristically similar pigments with different chemical structures. Such pigments frequently respond differently to the source of radiation (daylight or xenon arc lamp), irrespective of the surrounding vehicle system. The following studies demonstrate this very clearly. In letterpress proof prints and in long-oil alkyd vehicles, Pigment Yellow 17 is more lightfast than Pigment Yellow 13 if both are exposed to daylight. A xenon lamp, however, corrected with filters, reverses the results, and it is now Pigment Yellow 13 that is more stable. Pigment Red 57:1 and Pigment Red 184 react in the same manner. This behaviour reflects a difference in the spectral sensitivity of pigments, the effects of which are observed especially in the UV range and in the difference between natural daylight and artificial light from the xenon tubes (Figure 1.45).
Test specimens can only be compared in terms of lightfastness or weatherfastness if all samples show the same depth of shade. Up until a few years ago, such determinations were carried out at equal pigment concentrations.
1.6.6.3 Factors Determining the Lightfastness
The lightfastness of a pigment–vehicle system is a function of the:
- vehicle,
- substrate,
- pigment volume concentration,
- thickness of the layer,
- additives.
1.6.6.3.1 Vehicle
Comparative studies show the considerable impact attributed to the medium. Certain types of fat dyes, for instance, have lightfastness equal to step 8 on the ISO Scale (Blue Scale) if they are incorporated in polystyrene or similar polymers, while in other media their lightfastness drops to step 1 or 2.
Similar observations have been made on offset printing inks containing 15% Pigment Yellow 13 (Figure 1.46).

Figure 1.46 Effect of the binder on the lightfastness. Pigment content in the printing ink: 15% Pigment Yellow 13.
1.6.6.3.2 Substrate
To test for lightfastness with respect to the substrate, the same letterpress ink containing 20% Pigment Red 53:1 was printed on different papers (Figure 1.47). The first sample was printed on a standardized (DIN 16519) coated art paper without any optical brightener, the second substrate was a slightly fluorescent glazed magazine gravure paper, and the third was regular wood pulp paper for newsprint purposes, also slightly fluorescent.

Figure 1.47 Effect of the substrate (paper) on the lightfastness of letterpress proof prints. Pigmentation in the printing ink: 20% Pigment Red 53:1.
1.6.6.3.3 Pigment Volume Concentration
Lightfastness and weatherfastness of a pigmented system improve with the depth of shade; that is, the system becomes more stable as the pigment concentration increases, up to the critical pigment volume concentration. Since the pigment particles in the uppermost layer of the pigmented system are the first to be affected by incident light, increasing the pigment volume concentration in a layer can be expected to provide better fastness. This is clearly demonstrated by comparative studies on 1/3 and 1/25 SD letterpress prints containing Pigment Red 53:1 (Figure 1.48). The 1/25 SD prints, which contain much less pigment per cm2 than 1/3 SD prints and therefore provide a lower pigment surface concentration, fade much faster than 1/3 SD prints. The pigment volume concentration in a 1/3 SD print is much higher than that of a 1/25 SD print. The colour change upon irradiation, expressed according to DIN 6174 in CIELAB units, is shown in Figure 1.48.

Figure 1.48 Dependance of the lightfastness of letterpress proof prints with the standard depth of shade. Pigment Red 53:1, Curve A: 1/3 SD print; curve B: 1/25 SD print.
1.6.6.3.4 Effect of the Thickness of the Layer
Interestingly, despite equal composition and equal pigment surface concentration, two samples may respond differently to light, depending on how thickly they are applied. The effect of the film thickness has been experimentally demonstrated on a series of offset printing samples made from a 15% Pigment Yellow 12 formulation. Appropriately diluted with vehicle, this paste affords a set of inks, formulated at 12, 9, 6, and 3% pigment concentration, respectively. All inks are printed at the thickness required to match the original 15% pigment paste in terms of pigment surface concentration. Measurements are thus carried out on a series of prints that differ only in film thickness. Figure 1.49 shows that despite equal pigment surface concentration, the samples respond very differently to light. The effect of film thickness on the lightfastness of a print has not yet been explained mechanistically. The assumption is that the vehicle, which absorbs radiation in the longwave UV range and in the shortwave visible region, protects the pigment particles from incident flux. This protection improves with the thickness of the layer. Reducing the pigment volume concentration may be visualized as protecting the average pigment particle from incoming radiation by a thicker absorptive vehicle layer.

Figure 1.49 Influence of the pigment volume concentration on the lightfastness at constant pigment surface concentration of the resulting letterpress proof prints (different thickness of layers). Pigment concentration in the printing ink: 3, 6, 9, 12, 15% Pigment Yellow 12.
1.6.6.3.5 Effect of Additives
Since lightfastness and weatherfastness are properties of entire pigment–vehicle systems, additional components must be expected to have a more or less pronounced effect on the sensitivity of a material. In referring to test results, it is therefore necessary to mention the exact composition of the sample. In some systems, problems arise with peroxides, which may rapidly destroy organic pigments. UV absorbers, on the other hand, may improve the lightfastness of a system. The chemical process underlying the degradation and/or destruction of organic pigments through light or atmospheric conditions is difficult to elucidate, since they consist of a large range of different chemical compounds, each of which must be expected to react on an individual basis. Atmospheric contaminants such as peroxides, which appear as products of radiation, provide a certain amount of common ground in that they frequently initiate the degradation process. Since each pigment reacts according to its own specific mechanism, it might be useful to study the photochemical degradation mechanism of pigments, although to our knowledge organic pigments have not yet been examined individually.
Obviously, there are not only external factors but also the components of a pigmented system that largely define weatherfastness and lightfastness. The chemical and micromorphological degradation mechanisms in systems containing titanium dioxide, for instance, have been investigated extensively [184]. The results, especially those concerning micromorphological degradation of layers, may also be applied to organic pigment systems.
The electron photomicrographs in Figures 1.50–1.52 show the degradation of several pigment–vehicle systems in response to irradiation and weathering. These examples are borderline cases, in which either the pigment degrades considerably faster than its medium or the vehicle is the first to decompose.

Figure 1.50 Electron micrograph of an ultramicrotome-cut thin section of a nonirradiated gravure printed layer. Pigment concentration in the printing ink: 4% Pigment Yellow 12.

Figure 1.51 Electron micrograph of an ultramicrotome-cut thin section of an irradiated gravure printed layer. Pigment concentration in the printing ink: 4% Pigment Yellow 12.

Figure 1.52 Pigment Red 48:4 in an alkyd-melamine resin baking enamel. Electron micrographs of ultramicrotome-cut thin sections of an irradiated coating (above: detail).
Rapid pigment degradation relative to the vehicle is shown in Figures 1.50 and 1.51. These figures show ultrathin sections produced with an ultramicrotome. The sections were cut out of a gravure printed layer made from Pigment Yellow 12 and a vehicle based on a colophony-modified phenolic resin before and after irradiation. The decomposition of the pigment crystals in the irradiated sections is already far advanced: a large number of voids have replaced the pigment crystals. The degradation products apparently migrate into the vehicle. The chlorine content of these compounds makes them easy to recognize by the concentric black rings that surround each pigment particle, as shown in electron photomicrographs. A closer look shows that pigment destruction, visible through these black spots, is most advanced on the exposed side of the ink film. The sections that are closest to the surface are almost entirely black, indicating advanced pigment destruction. Moreover, the vehicle has started to decompose.
Figure 1.52 shows the surface of a corresponding P.R.48:4 coating (Section 2.7.2.4) made of an alkyd-melamine system. The samples were examined after a 5000 h exposure in an accelerated exposure unit (Xenotest 1200).
If, on the other hand, the vehicle is less stable to light and weather than the pigment, then lightfastness and weatherfastness of the entire system are largely a function of the vehicle sensitivity. Figure 1.53 shows electron photomicrographs of ultrathin sections of coatings containing pigments embedded in an alkyd-melamine system, taken before and after exposure; the sections were cut with an ultramicrotome. Pigment crystals of unsubstituted γ-quinacridone are beginning to protrude from the surface of the coating. These are the particles that, being more lightfast, have survived the decomposition of the surrounding vehicle; they can be removed by polishing or rubbing.

Figure 1.53 Electron micrographs of ultramicrotome-cut thin sections of an irradiated (a) and a nonirradiated (b) layer (alkyd-melamine resin baking enamel) containing Pigment Violet 19, γ-modification.
1.6.7 Thermal Stability
The fact that not all pigments are thermally stable considerably limits the choice of products that are useful in systems processed at high temperatures. Thermal stability is a particular concern in connection with the spin dyeing of polypropylene, polyester and polyamides (Section 1.8.3.8), and the mass colouration of low-density polyethylene and polypropylene (Section 1.8.3.1). The maximum temperature to which an organic pigment is subjected during such a process is between 260 and 320 °C, and the pigments are usually exposed for a few seconds to minutes only.
Few organic pigments resist these heating conditions. As a result, there are some shades that are inaccessible to organic pigments with such thermal requirements.
Insufficient thermal stability may affect the technical properties of a pigment, particularly its colouristics and fastness properties.
Thermal stability is always a system-dependent property. It is a function not only of the chemical composition of the medium but also of the processing conditions, degree of dispersion and pigment concentration. For most practical purposes, for instance, the colour change due to pigment decomposition in a highly pigmented system is hardly noticeable and can thus be tolerated.
Colour change in a pigment–vehicle system may originate from the following phenomena [185]:
- Thermal pigment decomposition
Degradation occurs if a pigment–vehicle system is processed above the decomposition temperature of the pigment. Pigments incorporated in a solid or melt sometimes decompose spontaneously by undergoing a reaction that is initiated at a distinct temperature. In these cases, differential thermal analysis may be applied to detect the thermal transitions associated with pigment decomposition (Figure 1.54). Such abrupt changes, however, are rare. More commonly, pigment degradation is a subtle transition. Polycyclic pigments in particular undergo a continuous, slow exothermic process over a wide temperature range (Figure 1.55). The thermographs, however, do not differentiate between pigment degradation and slow physical alterations of the pigment particles, such as phase transitions (change of crystal modification) or changes in the particle size.
- Chemical interaction between pigment and application medium
The heat sensitivity of a system is rarely affected by reactions between pigment and vehicle, but two examples should be mentioned. As a powder coating is cured, the organic pigment may start to interact with other components in the system [186]. Pigment Red 168, Dibromoanthanthrone (Section 3.7.4.2), incorporated in an epoxy resin powder, for instance, will react with a basic hardener such as dicyandiamide. The reflection curves in Figure 1.56 indicate that the system undergoes considerable colouristic changes.
However, the system is fast to acidic hardeners, such as trimellitic acid. The colouristic changes are accompanied by drastically reduced lightfastness. A certain dullness of shade, sometimes observed at higher curing temperatures, vanishes rapidly upon exposure to light; and the lightfastness of the resulting product equals that of the pigment component. Even under mild conditions, the bromine atoms in the anthanthrone molecule will be replaced by amino or cyano groups, resulting in grey to greyish blue pigments of poor lightfastness. Light exposure and other tests indicate that the pigment molecules that are closest to the surface of the material may interact with the hardener. It is very likely that, under curing conditions, the pigment molecules undergo a substitution reaction, exchanging bromine atoms by cyano groups, which results in dull compounds with reduced lightfastness. The substitution products are thought to be responsible for the observed changes in the colouristics and the fastness properties of a pigment–vehicle system.
Special consideration should be paid to metal complexes such as hydrazone methine pigments (Section 4.2). At high temperatures, the yellow copper complex with the chemical constitution 10, incorporated in PVC, will exchange its chelated copper atoms with the metal atoms present in the application medium. Stabilizers containing barium/cadmium or lead produce yellow shades, while dibutyltin thioglycolate or other tin compounds produce a brilliant medium red. Colour change is slow at low temperatures, but at 160 °C the effect is rapid [185].

- Dissolution in the application medium
A certain degree of pigment solubility in its medium is frequently responsible not only for poor heat fastness but also for processing problems, such as bleeding (Section 1.6.3.2) or blooming (Section 1.6.3.1). A pigment that partially dissolves in its medium must be expected to change colour (Figure 1.57) [117]. Transmission curves for Pigment Red 170 (Section 2.6.4), dissolved in dimethylformamide or suspended in water, indicate the colouristic behaviour of the system. Colouristic changes make it easy to monitor dissolution effects. Thermal stability frequently parallels the fastness of a pigment to migration (Table 1.3) [185]. The investigation was carried out on several hydrazone pigments, incorporated in an alkyd/melamine resin paint reduced 1:20 with TiO2. Thermal stability was established by baking the samples at 120 and at 200 °C. The resulting colouristic changes were compared to known data on the migratory tendencies of each pigment in the test system (overpainting fastness) and its fastness to bleeding in plasticized PVC, determined according to DIN EN 14469. Colour changes are referred to in terms of CIELAB units. The results clearly indicate that solubility in these pigments parallels thermal stability.
- Changes in the physical characteristics and particle size of a pigment
Elevated temperatures tend to modify the crystal structure of a pigment. Characteristic changes in the X-ray diffraction pattern include line sharpening, that is, decreased half-width, while the relative intensities of the characteristic diffraction lines typically increase, indicating enhanced crystallinity and a higher portion of large-sized crystals. Analysis of the scattering pattern shows that there are now larger coherent areas that appear as units under X-ray (Section 1.5.4).
These are the changes that are observed in a pigment powder as it is processed or heated in the application medium. Modifications in the sizes of the crystallites of a polycyclic pigment treated in its application medium under varying conditions [185] may be derived from X-ray crystallographic data. Table 1.4 reflects a clear correlation between crystallinity and thermal stability: increased crystallinity leads to improved heat fastness.
Polymorphous pigments, on the other hand, may undergo phase transitions as the temperature rises (Section 1.5.3), indicating that different crystal modifications respond differently to heat.
Several thermal stability tests are available, some of which have developed into national (DIN) or international industrial standards (ISO). Pigments in thermoplastic systems, for instance, are studied under heat extrusion conditions [187]. The colourant to be tested, possibly together with titanium dioxide, is dispersed in the thermoplastic, using a mixer and a granulating extruder (Section 1.8.3). The pigmented test pellets are then fed into a screw extruder, which ejects a standardized test specimen with defined dimensions [188]. Starting at the lowest possible temperature level, the extrusion temperature is increased by intervals of 10 or 20 °C between samples.
To assess the colour change of the test films, each sample is compared colourimetrically with the standard, which is processed at the lowest temperature. The thermal stability of the colourant in the test medium is defined by the interpolated temperature value at which the colour difference between sample and standard equals ΔE*ab = 3. Determinations are carried out at various Standard Depths of Shade; common values are 1/3 and 1/25.
Pigments incorporated in plasticized or unplasticized PVC are assessed by different methods [189, 190]. The preparation of test dispersions is commonly performed on a standardized two-roll mill at temperatures between 190 and 195 °C or, respectively, between 180 and 185 °C. Samples are taken at intervals of 10, 20 and 30 min, moulded into films and evaluated for colour change.

Figure 1.54 Thermal decomposition of hydrazone pigments in the solid phase. Results of differential thermoanalysis performed on pigment powders in a nitrogen atmosphere.

Figure 1.55 Thermal decomposition of polycyclic pigments in the solid phase. Results of differential thermoanalysis performed on pigment powders in a nitrogen atmosphere.

Figure 1.56 Effect of the hardening system in an epoxide resin powder coating on the colouristic properties of the paint. Pigment: Dibromoanthanthrone (Pigment Red 168). Crosslinking conditions: 15 min at 180 °C. Curve 1: trimellitic anhydride, curve 2: imidazoline hardener and curve 3: accelerated dicyandiamide hardener.

Figure 1.57 Transmission spectrum of Pigment Red170 dissolved in dimethylformamide or in an aqueous suspension.
Table 1.3 Relation between the heat stability of hydrazone pigments in an alkyd-melamine resin baked system, their fastness to overpainting in this paint system and their bleed resistance in plasticized PVC.
| Colour Index Name | Temperature effect in CIELAB unitsa | Fastness to overpainting on a fastness scale from 1 to 5 | Bleed resistance on a fastness scale from 1 to 5 |
| P.R.112 | 28.1 | 1 | 1 |
| P.R.3 | 12.7 | 2 | 1 |
| P.Y.1 | 10.9 | 2 | 1 |
| P.Y.74 | 4.5 | 3 | 1 |
| P.Y.12 | 4.4 | 4–5 | 1–2 |
| P.R.170 | 3.1 | 4 | 2–3 |
| P.Y.83 | 1.9 | 5 | 4–5 |
| P.O.36 | 1.3 | 5 | 5 |
| P.R.175 | 0.5 | 5 | 5 |
| P.R.144 | 0.2 | 5 | 5 |
| a) Colour difference between a coating baked at 200 °C and one baked at 120 °C. (Reduced 1:20 with TiO2.) | |||
Table 1.4 Changes in the crystallite size of some polycyclic pigments in coatings or plastics colouration as a result of heat exposure (calculated from the X-ray powder diffractograms).
| Pigment | Temperature exposure | Size of crystallites (Å) |
| P.R.123 | Baked 5% | |
| In full shade | ||
| In alkyd/melamin resin paint: | ||
| 30 min, 120 °C | 225 | |
| 30 min, 240 °C | 340 | |
| P.R.122 | Iaked 5% | |
| In full shade | ||
| In polyester-modified silicone resin system: | ||
| 60 min, 200 °C | 245 | |
| 60 min, 300 °C | 295 | |
| P.R.149 | Coloured in | |
| ⅓ Standard depth of shade in low pressure polyethylene: | ||
| 5 min, 200 °C | 270 | |
| 5 min, 300 °C | 380 |
1.6.8 Flow Properties of Pigmented Systems
1.6.8.1 Rheological Properties
Newton's law states that for a liquid under shear, the shear stress (τ) is proportional to the shear rate. In this sense, most of the unpigmented vehicles used in the paint and printing ink industries are considered ideal or Newtonian liquids. The ratio of the shear stress (τ) to the shear rate (D) is thus a constant (η), dependent only on temperature and pressure. This is not true for specialized gel varnishes and thixotropic systems, which are designed to have special rheological properties.
Ideal and non-Newtonian behaviour is demonstrated by the flow curves in Figure 1.58, in which the shear rate is plotted versus the shear stress.

Figure 1.58 Typical flow curves.
The first example describes a Newtonian fluid, represented by a linear correlation whose slope equals the viscosity of the material. When a pigment is dispersed in a vehicle, the behaviour of the resulting liquid deviates more or less from Newtonian flow. The previously linear shear stress–shear rate function is transformed into a curve whose slope decreases with increasing shear rate and shear stress. This phenomenon is known as structural viscosity or pseudoplastic behaviour. Shearing is thought to break up the pattern of pigment–vehicle interaction, deforming or rearranging the units and lining up the previously disordered vehicle molecules, thus reducing their effective cross-section and affording reduced viscosities. This reversible process is not time-dependent, that is, there is a definitive correlation between shear stress and viscosity, no matter how long the stress is applied. Several such systems have been studied empirically and their behaviour has been described by flow curves. For practical purposes, the Casson flow model is a useful approximation to describe the structural viscosity of a pigment–vehicle system. It is expressed by the following equation:
A Casson fluid is rheologically identified by two parameters: yield value and plastic viscosity. The plastic viscosity relates to the asymmetry of the flow particles and the yield value is connected with the forces of attraction between particles. The plastic or ultimate viscosity (η∞) in pascal seconds (Pa·s) may be determined from the slope of a shear stress versus shear rate plot, as the shear rate tends to infinity. The yield value τo (in N cm−2), on the other hand, is determined by extrapolation to D = 0.
The ultimate viscosity (η∞) of a pigmented medium is practically independent of the particle size, but increases appreciably with rising pigment volume concentration and higher vehicle viscosity [200].
Figure 1.59 quantifies the relation between the yield value (τ∞) and several system-inherent parameters. The curves show that τ∞ decreases with the cube of the particle size and increases drastically with the pigment volume concentration. Since the yield value is an indication of pigment–vehicle interaction, it is also proportional to the degree of pigment dispersion [191]. Interparticle attractions have received considerable theoretical and experimental treatment, and numerous original publications and reviews are available (such as References [192–194]).
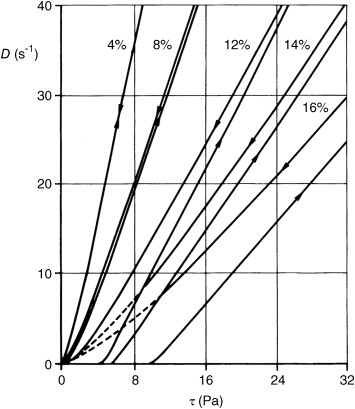
Figure 1.59 Influence of the pigment concentration on the rheological behaviour. P.Y.73 in air drying alkyd resin paint.
Thixotropy is one of the reversible time-dependent effects that constitute nonideal behaviour (Figure 1.59).
An increasing pigment concentration is associated with more or less pronounced thixotropy (Figures 1.58 and 1.59). Thixotropic systems have a gel structure. Shear, however, breaks up the previously stable pigment–vehicle structures. The viscosity of a thixotropic fluid therefore decreases with time when shear is applied, even at constant shear rate, and finally approaches a minimum. The graphs in Figure 1.59, read from left to right, exemplify the occurrence of thixotropy with increasing pigment concentration in a paint system. When the shearing action stops, the original gel structure reappears after a certain period of time.
Thixotropic decay also depends on the shear rate. At a given shear rate D (s−1), an equilibrium is reached that does not normally coincide with the sol value. As mentioned above, thixotropy may thus be described as a time-dependent and reversible change in the consistency of a material, observed during and after the effect of shear. Several processes are affected by the rate at which the matrix within a system is built up and the rate at which it is broken down with shear. Examples include the time needed for a dispersion to flow out of a ball mill or other dispersion unit and separate from the milling medium at different points in time after completion of the dispersion process. Thixotropy plays an important role at various steps throughout the printing process, such as colour separation, and provides the basis for thixotropic paints and inks. Figure 1.60 illustrates the time that is necessary for the thixotropic structures of two systems to rebuild on standing (Section 1.6.5.8).

Figure 1.60 Rate of thixotropy buildup. (a) Paste A shows a continuous buildup over time. (b) In paste B, buildup of the structure seems to be already completed after a few minutes; however, considerable change is still observed 16 h later.
Dilatancy occurs less frequently in pigmented systems. In flush pastes or pigment concentrates, which are formulated at high pigment levels, shearing may produce an increase in viscosity (η). As the pigment concentration reaches the vicinity of the critical pigment volume concentration, and even more so if this point is exceeded, increasing the shear stress (τ) or the shear rate (D) will thicken the fluid (Figure 1.58). This may create a problem with aqueous pigment preparations, which are sometimes automatically metered into the application system under considerable shear. These pastes should therefore be tested for dilatancy at high shear rates before they are processed.
Rheopexy, a reversible time-dependent effect like thixotropy, is a rare phenomenon in pigmented systems. Rheopectic fluids increase in viscosity (η) with time when sheared at a constant shear rate (D) or a constant shear stress (τ) until they approach a viscosity maximum (Figure 1.58).
1.6.8.2 Viscoelastic Properties
Most pigmented systems are considered viscoelastic. At low shear rates and slow deformation, these systems are largely viscous. As the rate of deformation or shear rate increases, however, the viscous response cannot keep up, and the elasticity of the material increases. There is a certain amount of emphasis on viscoelastic behaviour in connection with pigment dispersion as well as ink transportation and transformation processes in high-speed printing machines (see below). Under periodic strain, a viscoelastic material will behave as an elastic solid if the time scale of the experiment approaches the time required for the system to respond, that is the relaxation time. Elastic response can be visualized as a failure of the material to flow quickly enough to keep up with extremely short and fast stress/strain periods.
The viscoelastic behaviour of a printing ink or other material is largely a function of the polarity of pigment and medium [195]. However, it is difficult to quantify the complex rheology of a viscoelastic system, because structural viscosity, thixotropy and dilatancy may all occur at the same time.
1.6.8.3 Influence on the Flow Properties
Rheology control is necessary in connection with the manufacture, handling and application of pigmented systems; however, the requirements of today's industry are so complex that only a few aspects can be discussed within the scope of this book. Flow is defined by various parameters. Pigment-determined factors include concentration (Figure 1.59), specific surface area (Section 1.7.8), particle shape and surface structure. The system also plays a major role. Flow is a function of the choice of components and composition of the entire system, interactions between the various components and between components and pigment surface, dispersion conditions, and the conditions that govern various stages during the application. Amongst these aspects, the emphasis is on the dispersion conditions of the pigment (Sections 1.6.5 and 1.7.5). The operation and effectivity of the dispersion equipment [149] as well as operational parameters have a considerable impact on the various facets of dispersion, especially concerning the breakup of the pigment agglomerates and wetting by the application medium. The degree of dispersion determines the surface area that is accessible to wetting by the various components in the system. Interactions at the interfaces between pigment and medium or between pigment and solvent also appreciably influence the response of a material to mechanical force.
1.6.8.4 Correlation between Flow Behaviour and Rheological Parameters
Although the rheology largely determines the application properties of a pigment in its vehicle system, the exact correlation between rheological and performance parameters remains elusive. Both quality control and research face problems because of the difficulty of predicting the behaviour of a pigmented system or systematically developing materials with certain defined flow properties. There are no standardized techniques to measure the rheological performance of a material under the conditions of practical use. To characterize a fluid, specialized instruments are used to simulate specific steps in the processing and application of a pigment–vehicle system. The printing inks industry, for instance, typically resorts to tackmeters [196]. The tack [197] or, its reverse, the shortness are primary applicational factors in determining the printability and therefore the commercial fate of an offset printing ink. Defined in 1957 [198], the equation:
relates to the stepwise change of one variable only. A test series in which either the pigment concentration or the composition of the medium is changed systematically correlates closely with the results of a test that is carried out under actual application conditions (Table 1.5) [199].
Table 1.5 Relation between rheological parameters and properties related to the printing of offset inks.
| Ink no. | τo × 105 (Pa) | η∞ (Pa·s) | Shortness (according to Zettlemoyer) | Printing test |
| 1 | 9.2 | 13.5 | 680 | Too long |
| 2 | 7.2 | 10.0 | 720 | Very long |
| 3 | 6.2 | 7.4 | 835 | Long |
| 4 | 4.8 | 5.3 | 905 | Long |
| 5 | 4.5 | 3.3 | 1360 | Short |
| 6 | 4.0 | 2.8 | 1430 | Too short |
However, the equation is medium-specific, and errors may occur if it is used to extrapolate data outside the range of experimental determination. Tackmeters simulate the conditions within the printing presses by rotating a test offset blanket cylinder versus an inked plate cylinder at different speeds. The generated force is measured by the deflection of the cylinder as it is pulled by the ink. The test results usually correlate well with the demands of practical offset printing.
The complicated rheology of a pigmented system under processing conditions is reflected in the flow behaviour of publication gravure printing inks in high-speed printing machines, especially in the gap between doctor blade and printing cylinder [200]. Studies carried out on modern high pressure capillary viscometers [201] indicate that the flow curves may show unexpected deviations at high shear rates. This is especially true if the test parameters (D = 106 s−1 and flow times t of up to 10−4 s) closely approach actual printing conditions (D = 107 s−1 and t = 10−6 s). Moreover, flow behaviour as a function of the shear rate (D) may differ between inks, as shown by a study comparing the flow curves of two publication gravure printing inks at D = 10 s−1 and at D = 104 s−1. The ink that displays higher viscosity at a low shear rate loses viscosity as the shear increases to 104 s−1, and the relation is reversed again at D = 105 s−1. The viscosity profiles thus cross each other and intersect at medium shear rates, at which both inks exhibit identical flow behaviour. Such unpredictable shear effects make it necessary to collect a complete set of data points to draw up system-specific flow curves for publication gravure inks. There is little point in using a low shear rate viscometer to determine the behaviour of a gravure printing ink that is to be processed at a high shear rate, since the flow curve cannot be extrapolated to higher values. Traditional viscometers or flow cups are inadequate even for qualitative purposes.
Publication gravure printing inks apparently show elastic behaviour; at D = 105 s−1, the modulus of shearing (G) is 103 Pa. This indicates that at high shear rates (D), between 105 and 106 s−1, gravure printing inks develop normal stress effects and thus show a hydrodynamic lubricating effect. These deductions are relevant to an understanding of the mechanical effects between doctor blade and cylinder. At D = 10 s−1 and an experimental time t = ∞, highly thixotropic gravure printing inks have an apparent viscosity (η) of 0.1 Pa·s; while at D = 105 s−1 and t = 10−3 s, the viscosity assumes values between 0.05 and 0.06 Pa·s.
1.6.8.5 Rheological Measurements
Numerous different commercial viscometers, which provide various geometries and a range of viscosity and shear rates, can be used to characterize a fluid. Specialized techniques have been developed to suit specific purposes [202–204].
Standards have been developed regulating the use of some of these commercially available viscometers. Such standards, for instance, cover the use of rotational viscometers with coaxial cylinders [205] if they are employed to measure flow curves and viscosities at shear rates between 5 and 1000 s−1. Systematic errors distorting viscosity determinations on Newtonian liquids using such rotational viscometers and corrections that have to be applied to the resulting measurements are discussed in the literature [206]. Other standards cover the testing of paints, which have to be measured at very high shear rates in the range between 5000 and 20 000 s−1 to reflect the conditions of actual paint application. These are tested according to industrial standards using rotational viscometers or standard cone-and-plate instruments [207]. Tests have been developed for letterpress and offset printing inks and similar systems that employ specialized falling rod viscometers [208]. These devices do not afford definitive flow curves for thixotropic systems but instead trace a line somewhere between the gel and the sol curve. There is extensive literature on techniques describing how to determine the rheology of special systems.
The determination of flow properties taken during the pigmentation and processing of thermoplastics are described in the literature on polymers [209, 210].
Since the rheology of many systems depends largely on the temperature, accurate and reproducible measurements require very careful temperature control. A 1 °C temperature drop, for instance, increases the apparent viscosity (η) of an offset printing ink by approximately 15%. To demonstrate the correlation between thixotropy and temperature, Figures 1.61 and 1.62 show the flow curves at different temperatures for two offset printing inks [211]. Both materials clearly lose thixotropy – indicated by the area under the thixotropic loop – as the temperature increases. This effect is much more pronounced in the first case (Figure 1.61), while the second ink exhibits a very slow decrease thixotropic behaviour (Figure 1.62).

Figure 1.61 Variation of thixotropy with temperature. Example of a printing ink whose thixotropy decreases rapidly with the temperature.

Figure 1.62 Variation of thixotropy with temperature. Example of a printing ink whose thixotropy decreases slowly with the temperature.
Thixotropy is frequently calculated by determining the area between the flow curves for increasing and decreasing shear rates. Standard procedures to establish this loop involve subjecting the tempered printing ink in a cone-plate viscometer to maximum tensile shear rates. After allowing the sample to rebuild its structure on standing at zero shear rate for 5 min, the up curve is determined by increasing the shear rate from 0 to maximum (300 s−1) within 5 s and shearing the sample at this rate for 30 s. The down curve results from reducing the shear rate back to 0 within another 5 s. It is important to remember that the exact shape of the flow curve for a given material depends on the history of the sample and the experimental procedure, for example the area under the curve characterizes the specific thixotropic properties in each case.
The flow behaviour of a system may change significantly during the first few hours after the shearing action stops. This is particularly true for systems with a high pigment concentration. The plot in Figure 1.63, for instance, represents the viscosity–shear-time profile of a toluene-based gravure printing ink. The pigment concentration is 15% (Pigment Yellow 12).

Figure 1.63 Variation of the thixotropy of publication gravure printing inks containing 15% Pigment Yellow 12 with the storage time after dispersion.
The viscosity drops at first and then increases as soon as the shearing action stops. This behaviour is attributed to superimposed reagglomeration effects (Section 1.7.5). These effects include reduction of the wetted surface area of the pigment particles and delayed wetting effects, which increase the viscosity of the ink. Easily dispersible pigments with good wettability exhibit no such time-dependent flow behaviour.
No time-dependent change of the rheological values is observed if the printing ink is produced at higher dispersion temperatures. Studies have been carried out on offset printing inks containing Pigment Yellow 13 that were dispersed at temperatures of 15 and 50 °C, respectively. Measurements were taken 4 and 24 h after dispersion (Figure 1.64). The effect of delayed wetting is negligible in the ink produced at 50 °C, because the surface of the pigment particles is wetted thoroughly at this high temperature.

Figure 1.64 Influence of the dispersion temperature ((a) 15 and (b) 50 °C) and the storage time on the viscosity of sheet offset printing inks containing Pigment Yellow 13. Measurement temperature 23 °C.
1.7 Particle Size Distribution and Application Properties of Pigmented Media
The application properties of a pigment–vehicle system depend largely on the particle size distribution of the pigment. This seemingly straightforward correlation would make it easy to predict the application properties of an entire pigment–vehicle system from pigment powder data if it was not for the fact that the particle size of a pigment in its medium is not normally the same as that in the powder (Section 1.5.2). It is, therefore, difficult to gain more than qualitative information about the final properties, unless the particle size distribution is determined directly on the dispersed pigment in its medium of application.
1.7.1 Tinctorial Strength
Figure 1.65 reflects the relation between pigment particle size and the ability of a pigment–vehicle system to absorb visible electromagnetic radiation, which correlates on the other hand to the tinctorial strength of a pigment in its application medium (Section 1.6.1.1). The ability of a given pigment to absorb light (its tinctorial strength) increases with decreasing particle diameter and accordingly increased surface area, until it approaches the point at which the particles are entirely translucent to incident light. Particle size reduction beyond this point does not improve the tinctorial strength of a pigmented system [212]. The tinctorial strength increases within the above-mentioned range of particle sizes, but only if the self-scattering of the pigment, for instance in white reductions with TiO2, can be neglected. Unusual optical characteristics may also be found in transparent systems such as printed layers and may change the relation between tinctorial strength and particle size distribution.

Figure 1.65 Relationship between particle size and absorption (tinctorial strength) of a pigment.
To study the effect of changes in the particle size or particle size distribution on the optical properties of a TiO2-reduced pigment–vehicle system, a series of aqueous Pigment Yellow 1 pastes in emulsion paints were compared [213, 214]. Ultracentrifugation techniques were used to separate the pigment pastes into three fractions according to the particle size. Figure 1.66 shows the electron photomicrographs of the individual fractions; the respective particle size distributions as determined by ultrasedimentation are outlined in Figure 1.67, in which the median for each curve (Section 1.5.2) is indicated by an arrow. Each fraction was then blended with an appropriate TiO2 dispersion and the resulting samples matched against the original batches. At equal reduction ratio, the tinctorial strength of 1% colourations of the medium sized particle fraction (paste 2) matches that of 0.7% colourations of the batch with a fine particle size (paste 3) and the tinctorial strength of 2.1% colourations of the product with a coarse particle size (paste 1). The results can be interpreted quantitatively: the tinctorial strength of the three fractions in the order of increasing particle size thus corresponds to a pigment/tinctorial strength ratio of 100 : 145 : 300.
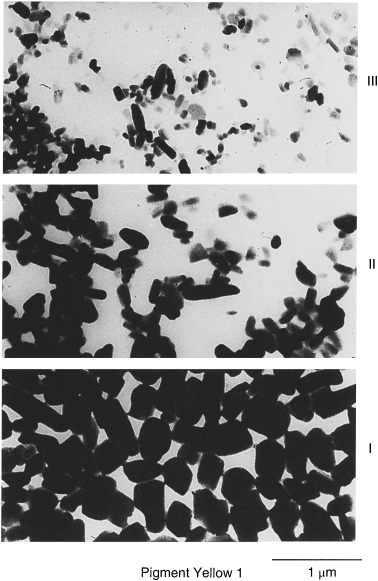
Figure 1.66 Electron micrographs of samples I–III, respectively.

Figure 1.67 Particle size distributions of the aqueous preparation of Pigment Yellow 1 samples I–III, respectively (see Figure 1.66).
There are several specific cases in which, independent of the application, the tinctorial strength of a product is even more sensitive to the particle size distribution.
A comparative study of three letterpress proof prints containing Pigment Yellow 13 also illustrates the correlation between particle size distribution and the tinctorial strength of prints. The electron photomicrographs of the three samples in Figure 1.68 correspond to the particle size distribution histograms in Figure 1.69. All three pigments were dispersed under equal conditions in a sheet offset vehicle using a three-roll mill; and printed 1 µm thick with a proofing printer. Colourimetric evaluation shows that compared to sample III, which contains the variety with a coarse particle size, sample II shows a 25% increase in strength; while the print with a fine particle size exceeds the standard with a coarse particle size by as much as 36%.

Figure 1.68 Elecron micrographs of Pigment Yellow 13 samples of different particle sizes.

Figure 1.69 Particle size distributions of three samples of Pigment Yellow 13 (see Figure 1.68).
1.7.2 Hue
The shade of a product is another parameter that is influenced by the particle size (Section 1.6.1.2). This is easy to see by comparing the reflection curves of two Pigment Yellow 83 white reductions in an alkyd-melamine resin system. The electron photomicrographs in Figure 1.70 and the particle size distribution histograms in Figure 1.71 represent these samples. At equal pigment concentration, curve 1 of the two remission curves in Figure 1.71 reflects the behaviour of the pigment with the smaller particle size, and curve 2 that of the type with the larger particle size. In the wavelength region between about 520 and 560 nm, curve 2 undergoes a sideways shift, indicating a distinct change of shade towards a more reddish yellow.

Figure 1.70 Electron micrograph images of two samples of Pigment Yellow 83 of different particle sizes.

Figure 1.71 Particle size distributions of two P.Y.83 grades (see Figure 1.70).
In general, the following rule applies: increasing particle size causes yellow pigments to undergo a redshift. Orange pigments turn more reddish, yellowish reds become more bluish, bluish reds turn more yellowish, brown shades undergo a redshift, violet pigments, such as Dioxazine Violet, develop a more bluish hue, and blues, such as phthalocyanine pigments, show more of a reddish shade. The influence of particle size on the shade is especially evident in white reductions.
Paints behave similarly. Figure 1.72 illustrates the colour shifts observed in Pigment Yellow 1 pastes containing different particle size distributions (Section 1.7.1). The colours are identified according to their location on the DIN Colour Chart, in which each colour is defined by the two tristimulus values, x and y [215]. Increasing amounts of pigment paste were added to the sample of a pure white dispersion, referred to by location W. Likewise, the shades of the differently sized Pigment Red 3 (Toluidine Red) dispersions were plotted within the same area of the Colour Chart [213]. All locations along one of the numbered lines originating in point C indicate equal shade, while the concentric rings are lines of equal colour saturation. It is not difficult to recognize that at constant pigment concentration the varieties with a coarser particle size of the two pigments differ in their tinctorial behaviour relative to the respective type with a fine particle size. The Pigment Yellow 1 with a coarse particle size sample tends more towards the reddish end of the scale, while its Toluidine Red counterpart shifts clearly toward the bluish region. Similar colour shifts are observed if the amount of pigment in the dispersion is increased, a tendency that is most pronounced in the respective samples with a fine particle size.

Figure 1.72 Colour locations of aqueous Pigment Yellow 1 and Pigment Red 3 pastes of different particle sizes. Section of the xy-chromaticity diagram, including the mid-point, source C (DIN 6164).
It has often been observed that the colouristic properties of an organic pigment are a function not only of the size of particles but also of their shape. This is due to the anisotropy of the optical properties in different crystallographic directions within the crystal forms of a pigment. In 1974 [216, 217], it was demonstrated that of the equally sized but differently shaped particles of beta copper phthalocyanine blue, the almost completely cubic, that is, more or less isometric, form produces greenish blue shades, while acicular forms are responsible for reddish blue hues. The optical behaviour of ordered pigment particles in systems has been reported in the literature [218, 219].
1.7.3 Hiding Power, Transparency
The hiding power of a pigment – a feature that is controlled by parameters such as the absorption coefficient of the pigment, the relative refractive indices of pigment and vehicle, the degree of pigment dispersion, the thickness of the layer, and other factors – is also largely determined by the scattering coefficient of the pigment. Like absorption, this coefficient is in turn a function of the particle size. The curve in Figure 1.73, plotting the scattering coefficient versus the particle size, goes through a maximum. Particles above this critical size exceed the scattering maximum only to lose opacity, a phenomenon known as ultratransparency [220]. Interestingly, the scattering maximum is frequently observed at a particle size that provides comparatively poor absorption and inadequate tinctorial strength.

Figure 1.73 Relation between particle size and scattering ability of a pigment.
A pigment that absorbs a large portion of light will increase the hiding power of its application system despite insufficient scattering. In highly concentrated formulations, coloured organic pigments with very high absorption coefficients and low scattering coefficients are referred to as hiding, even if the resulting full shade tends to appear somewhat dark. The excellent hiding power of copper phthalocyanine blue, for instance, is a result of a very large absorption coefficient, that is, high tinctorial strength [221].
However, there is no exact quantitative method that makes it possible to calculate the critical particle size that will produce an optimum in hiding power in a coloured pigment formulation. Although the basic theory behind this phenomenon has been treated extensively, it is still most advantageous for practical R & D purposes to experimentally determine the particle size that affords a maximum in hiding power. A more approximate than quantitative rule has been established by H.H. Weber, which makes it possible to estimate the particle size that will afford a maximum in scattering:
in which λ is the wavelength of the scattered light, and np and nB denote the refractive indices of pigment and vehicle, respectively. The refractive indices of organic pigments range between 1.3 and 2.5, dependent on pigment and wavelength, while most of the commonly used vehicles have an index somewhere near 1.5. Assuming, for example, that np = 2, then Weber's rule predicts that the scattering maximum for wavelengths between 400 and 700 nm will be realized by particles of 0.4–0.7 µm.
There are several techniques for determining the hiding power of a pigmented medium for practical application. Coloured systems are typically tested by measuring their ability to entirely cover up a standard black and white chequerboard substrate. Parameters used as variables are the pigment concentration and/or the thickness of the layer (pigment concentration per area unit), either one or both of which are adjusted until the ΔE*ab between the colours above the black and the white substrates equals 1 [222] (Section 1.6.3.1).
Using Pigment Orange 34 in a long-oil alkyd resin paint as an example, this technique clearly reveals the effect of the particle size distribution on the hiding power of the film. The two electron photomicrographs in Figure 1.74 reflect the structure of a transparent type with a fine particle size on the one hand (sample I) and a considerably more opaque type of the same pigment with a very coarse grain on the other hand (sample II).

Figure 1.74 Electron micrograph of Pigment Orange 34 samples of different particle sizes.
These correspond to the two different particle size distributions shown in Figure 1.75. Remarkably, two equal amounts of the same compound, incorporated into the same vehicle and applied at equal thickness, give an entirely different impression above a chequerboard surface (Figure 1.76).

Figure 1.75 Particle size distributions of Pigment Orange 34 samples I and II (see Figure 1.74).

Figure 1.76 Pigment Orange 34 samples of different particle sizes (above: small, below: large) in a long-oil alkyd resin system. Pigment concentration in the system: 6%; thickness of the wet film: 75 µm.
If the hiding power of a system is particle-size dependent, then the same is necessarily true for the transparency of the system. The criteria for satisfactory transparency in a pigment–vehicle system depend on the application purpose for which it is targeted. The printing industry, for instance, requires inks that do not appreciably cover black substrates and therefore uses formulations that scatter very little light. Printing inks are accordingly tested by applying standard layers of ink (1–2 µm) at certain pigment levels, respectively pigment surface concentrations, to black test paper and evaluate the results visually or colourimetrically.
The ability of scattering light, respectively the transparency of a pigmented layer can be determinded by measuring the tristimulus value y or the colour difference ΔE*ab according to DIN 6174 to compare increasingly thick layers. To quantitatively describe the transparency of a system, the so-called transparency number (T) has been introduced. It is defined as the inverse of the distance ΔE*ab between colours printed on top of a black surface and ideal black for a pigmented medium that has been applied in a layer of thickness h [223, 224]:
However, the transparency number is not only a function of pigment and vehicle; it usually changes with the thickness of the layer (e.g. the pigment surface concentration). The transparency number of a sample applied at a thickness somewhere in the interval between two limits h1 and h2 is calculated as follows:
It is not difficult, for instance, to find the respective transparencies of the two Pigment Orange 34 samples, which differ in their particle size distribution. In letterpress proof prints, printed 1.5 µm thick, 15% pigment formulations of both yield the following results:
| Transparency Number T | |
| Pigment sample 1 | 0.135 |
| Pigment sample 2 | 0.045 |
1.7.4 Lightfastness and Weatherfastness
The lightfastness and the weatherfastness of an organic pigment are largely dependent on the particle size and on its crystal structure, which is not surprising given that it is the interaction of pigment particles with radiation that determines their response to light. Very little is known about the chemistry responsible for the destruction of organic pigment crystals through incident light. In a pigment particle the molecules – unlike dissolved dyestuff molecules – are stabilized by the crystal lattice. This stabilization is caused by several effects:
- If a molecule is excited by a photon, radiationless decay of the excited state is likely, owing to the strong intermolecular interactions and the dense molecular packing in a pigment crystal.
- The degradation of a photochemically excited molecule starts in many instances with a change of the conformation (e.g. a rotation of fragments or a twisting of the molecule). In a crystal such conformational changes can be hindered by the lattice.
- If a bond is broken, the fragments cannot move apart; they stay in their positions, and may recombine.
- The radiation, which ultimately cleaves the pigment structure, only penetrates the pigment crystals by an average of 0.03–0.07 µm, which explains why, in larger particles, only the uppermost layers, those in the vicinity of the surface, are affected. The effect of light thus destroys one layer after another, beginning with the outer one. More centrally located molecules are safe until the outer layers are destroyed; therefore, large particles resist light much longer than small ones.
- In media such as plastics, UV light generates radicals (enhanced, for example, in the presence of titanium dioxide; but the presence of organic pigments alone may also cause radicals). These radicals migrate through the medium. Dissolved molecules are easily destroyed, while in pigment particles only the surface layer can be destroyed, leaving the interior of the particles unaffected.
- Oxygen and other reactive species that can react with dissolved molecules can hardly penetrate into the interior of pigment crystals, owing to the dense packing of strongly bonded molecules. Again, mainly the surface molecules are destroyed. Degraded molecules tend to stick to the surface, due to strong adsorption on the pigment surface.
Large particles, having a reduced surface-to-volume ratio and a reduced solubility, possess a higher light- and weatherfastness than small particles. Similarly, pigments that are slightly soluble generally have a lower light- and weatherfastness than pigments that are totally insoluble.
A few examples exemplify the relation between the particle size distribution of a pigment and its lightfastness. Figure 1.77 shows histograms of the respective particle size distributions of three aqueous pastes containing Pigment Orange 5 [225], determined by the Marshal ultrasedimentation method.

Figure 1.77 Particle size distributions of three Pigment Orange 5 samples.
Particle sizing is designed such that every two out of these three samples agree in one size-related aspect. Although the medians of samples 1 and 2 are almost equal, their size distributions have an entirely different shape. Pastes 1 and 3, on the other hand, show a similarly narrow distribution, but their medians are dissimilar. White emulsion paint was added to all three samples to afford equal depth of shade, and the samples were exposed to light. After 1500 h, the tinctorial strength of sample 1 was 72% of its original value, while samples 2 and 3 only retained 46% and 39%, respectively, of their tinctorial strength (Figure 1.78).

Figure 1.78 Relative tinctorial strength of irradiated dispersion layers containing Pigment Orange 5 of different particle sizes (white reductions) (see Figure 1.77).
It is interesting to see the difference in weatherfastness between two pigments as related to the particle size. Two types of Pigment Yellow 151 were incorporated in an alkyd-melamine baking enamel and examined by electron microscopy. The particle images are shown by the electron photomicrographs and the particle size distributions given in Figure 1.79. The coatings were exposed in a Xenotest 1200 type accelerated weathering cabinet [226]; cycles were 17/3 min (Section 1.6.6.2) (Figure 1.80). Figure 1.80 demonstrates that the type with the larger particle size shows significantly better weatherfastness than the type with the smaller particle size.


Figure 1.79 Electron micrographs and particle size distributions of P.Y.151 samples.

Figure 1.80 Effect of the particle size of P.Y.151 in alkyd-melamine resin systems on the weather-fastness. Organic pigment : TiO2 = 1 : 3.
1.7.5 Dispersibility
All dispersion conditions being equal, the dispersibility of a given organic pigment in a particular vehicle is largely controlled by its particle size distribution. However, the mechanisms underlying the relation between particle size and dispersibility are somewhat complex and not easy to study. Although the problem has been discussed in the literature, most publications fail to provide detailed information as to the degree of agglomeration, surface structure and pigment preparation. Some studies simplify the problem by standardizing the pigments through uniform finishing procedures in order to attain equal dispersibility and equal or similar surface structure [227, 228].
The dispersibility of a pigment depends on the number of surfaces and edges that can, through attractive surface forces, unite the particles into aggregates and agglomerates. These surface forces are a function of the particle size and size distribution. Theoretically, the larger the particles, the lower the surface forces, and the easier it is to disperse the agglomerates [229]. A wide particle size distribution considerably increases the number of points of adhesion, allowing small particles to ‘glue’ the large units together [230]. Simultaneously, the small particles fill in or partially occupy the gaps and pores. The inner surfaces of the agglomerates thus become less accessible to the surrounding medium, and so reduce the wetting rate (Section 1.6.5). In all particle size ranges, pigments with a comparatively narrow particle size distribution are therefore easier to disperse than corresponding types with a wider range of sizes.
The particle size of a pigment, however, has an impact not only on those steps within the dispersion process in which pigment agglomerates are broken down. It also affects retroactive processes, such as reagglomeration, flocculation and rubout effects [231]. Reagglomeration (Section 1.5) occurs if the pigment particles are not covered adequately by the binder. Flocculates (Section 1.5) are loose associations of particles that separate easily through low shear such as stirring. In contrast to agglomerates, flocculates are adequately wetted and the space inside them is filled with vehicle molecules. Although they are only loose units, flocculates adversely affect the colouristic properties of a pigment–vehicle system by decreasing its tinctorial strength and gloss. The tendency to flocculate is a property not only of a pigment but of an entire system. Since it is a result of interaction between the surface of the pigment particles and the surrounding medium, this phenomenon is particle-size dependent. Flocculation can be inhibited or even avoided by adding traces of antiflocculation agents to the application medium [232]. Likewise, these agents prevent pigment floating or flooding, and various agents are available to cope with other particle-size dependent failures that may be observed during the processing of a low-viscosity system [233]. Moreover, the particle size is also responsible for specking, seeding and similar phenomena frequently observed in paints.
Figure 1.81, which plots the depth of shade of two P.O.34 samples versus the time needed to disperse the pigment, represents the dispersibility curve of two types with different particle size distributions. The two samples of Pigment Orange 34 are characterized in Figures 1.74 and 1.75 (Section 1.7.3).

Figure 1.81 P.O.34 in a long-oil alkyd resin system. Influence of the particle size on dispersibility. Dispersion unit: paint shaker, organic pigment : TiO2 = 1 : 5. Sample 1: fine particle pigment, sample 2: coarse particle pigment.
1.7.6 Gloss
The gloss of a pigmented system is a function not only of the degree of pigment dispersion but also frequently of the particle size distribution (Section 1.6.5). This is a common phenomenon in very thin applications, such as in offset printing, which produces layers less than 1 µm thick.
The effect of the particle size on the gloss of a coating is readily reflected through the haze that is frequently observed in air-dried alkyd systems. This is a surface abnormality that develops with increasing drying time as a result of the contraction of the film volume. It is particularly pronounced in Pigment Red 3 types, where it is known as the Toluidine Red haze. Bluish toluidine red pigments, which on the average contain more coarse particles, are more prone to turn hazy than the yellowish types with finer particle sizes [234]. This phenomenon is illustrated below in Figures 1.82–1.86.
As an example, two samples of Toluidine Red with different particle size distributions (Figures 1.82 and 1.83) were dispersed in a long-oil alkyd system using an oscillating shaking machine (Paint Shaker) [235]. The paints were applied to a polished glass surface and the gloss values monitored to detect any developing haze. After drying for 16 days, both specimens were evaluated for surface gloss. The hazy type with a coarser particle size loses more than twice as much gloss as the type with a fine particle size, which is imperceptibly hazy (Figure 1.84). Scanning electron micrographs show the different surfaces of the two coatings after 1 day (Figure 1.85) and after 8 days (Figure 1.86).

Figure 1.82 Electron micrographs of Pigment Red 3 samples of different particle sizes.

Figure 1.83 Particle size distributions of P.R.3 samples I (fine particles) and II (coarse particles) (see Figure 1.82).

Figure 1.84 Influence of the gloss of air-dried alkyd resin systems containing Pigment Red 3 of different particle sizes with the drying time. Pigment content of the systems: 8%.

Figure 1.85 Scanning electron micrographs of the different surfaces of the two coatings containing Pigment Red 3 after 1 day: non-hazing and hazing sample (Figure 1.84).

Figure 1.86 Scanning electron micrographs of the different surfaces of the two coatings containing P.R.3 after 8 days: non-hazing and hazing sample (Figure 1.84).
Considerable shearing forces will break down the coarser-sized particles rather than the finer ones. Interestingly, the tendency of a pigmented system to become hazy decreases as the dispersion time increases, accompanied by improved gloss, colour shift towards more yellowish shades, enhanced colour strength in white reductions and increased viscosity [234].
Similar results have been reported for other types of products with coarse particle sizes, such as Pigment Yellow 1.
1.7.7 Solvent and Migration Fastness
The solubility of any crystalline material decreases as the particle size increases; the correlation is quantitatively described by the Ostwald equation [236]:
in which:
- c is the solubility of crystals with radius r,
- c∞ is the solubility of very large crystals,
- σ is the surface tension,
- V is the molecular volume of the crystal,
- R is the gas constant,
- T is the Kelvin temperature.
For several reasons, it is very difficult to quantitatively determine the solubility of an organic pigment as related to the particle size. F. Gläser [13] found that a particle diameter of 0.3 µm represents somewhat of a threshold value. The solubility of pigment crystals above this size essentially equals that of very large particles (c∞) and is therefore largely constant; in moving towards finer particle sizes, however, the solubility increases appreciably (Table 1.6).
Table 1.6 Solubility as a function of the particle size.
| r (µm) | 10 | 3 | 1 | 0.3 | 0.1 | 0.03 | 0.01 | 0.003 |
| α (r)a | 1.004 | 1.013 | 1.03 | 1.14 | 1.5 | 3.78 | 54.6 | 83 300 |
| a) α (r) is the factor by which the solubility of a particle with radius r increases compared to the solubility of a very large crystal. | ||||||||
The kinetics of the dissolution process obeys the Noyes and Whitney equation [237], revised by Nernst and Brunner:
in which:
- C∞ is the concentration of the saturated, evenly stirred solution,
- C is the respective concentration t minutes after the material begins to dissolve,
- O is the surface area,
- D is the diffusion constant,
- δ is the thickness of the interface.
According to this equation, the dissolution rate is proportional to the surface area (O) of the particles and to the solubility of the pigment. The dissolution rate decreases more and more as the concentration of the solution approaches saturation, that is, the limit of solubility.
Pigments are supplied with solubility tables determined by the manufacturer. The results are reported with respect to the test method described in Section 1.6.2.1. Although pigment dissolution is not normally very advanced in these tests, many samples distinctly bleed, irrespective of the particle size.
Table 1.7 lists the effect of several solvents on three chemically different pigment pairs as a function of the particle size; sample 1 is the type with the finer particle size. The specimens are rated according to the extent of pigment bleeding into the solvent. Grading is from 1 to 5, with 1 referring to considerable colouration of the solvent, while a completely colourfast sample is rated as 5. Solvent colouration is referred to relative to the Grey Scale for evaluation of bleeding [238].
Table 1.7 Solvent stability as a function of the particle size.
| Specific surface area (m2 g−1) | Ethanol | Toluene | Butyl acetate | Acetone | Dibutyl phthalate | |
| P.Y.1 | ||||||
| Sample 1 | 43 | 3 | 1 | 1 | 1 | 1 |
| Sample 2 | 16 | 3–4 | 3 | 3 | 3 | 3 |
| P.Y.12 | ||||||
| Sample 1 | 86 | 5 | 2 | 3–4 | 3 | 4 |
| Sample 2 | 34 | 5 | 3 | 4 | 4 | 4–5 |
| P.R.53:1 | ||||||
| Sample 1 | 43 | 3 | 4 | 4 | 3–4 | 4–5 |
| Sample 2 | 16 | 3 | 4–5 | 4–5 | 4 | 4 |
Another facet that is affected by the particle size of a pigment is its tendency to recrystallize, a direct result of poor solvent fastness (Section 1.6.5). This is illustrated by three samples that differ in terms of particle size distribution (Section 1.7.3). The samples were dispersed in an offset vehicle, divided into batches, stored at different temperatures for 8 h and, finally, printed on a black-coated test paper under standardized conditions. The transparency of each specimen shows the effect of the particle size on the occurrence of recrystallization during storage (Figure 1.87) (Section 1.6.5).

Figure 1.87 Effect of the particle size of Pigment Yellow 13 on its tendency to recrystallize in a sheet offset printing ink. The samples were stored for eight hours at different temperatures, printed in equally thick layers over a black background, and evaluated in relation to standard black. The particle size increases from Sample 1 to 3.
The tendency of a pigment to migrate is less sensitive to changes in the particle size (Sections 1.6.3.1 and 1.6.3.2).
1.7.8 Flow
Most of the flow behaviour and the rheological demands of a pigmented system are controlled essentially by the particle size of the dispersed pigment.
The correlation between rheological data and the most frequent particle diameter (Dmf) has been studied on a dispersion of the gamma modification of Pigment Violet 19 and various types of copper phthalocyanine blue in an offset vehicle on the basis of linseed oil [227]. The results show that while the ultimate viscosity (η∞) (Section 1.6.8.1) is independent of the particle size, the yield value increases with the cube of the particle diameter.
The considerable influence of the pigment particle size in controlling the flow behaviour and the rheology of a system is a useful asset to the manufacturer who tries to produce highly opaque paints. Comparatively large particles lead to an optimum in hiding power and consequently provide a relatively small surface area to be wetted. The resulting products exhibit good flow behaviour and are not usually very viscous. Larger amounts of such pigments can be worked into an application medium without affecting the rheology or the gloss of the system. To give a quantitative example, the following list gives the respective amounts of Pigment Yellow 74 types that, incorporated in a long-oil alkyd resin system, will afford essentially equal flow curves; the specimens differ in terms of particle size distribution and therefore vary in their opacity:
- 8.8 wt% pigment sample 1 (highly opaque type with coarse particle size);
- 5.6 wt% pigment sample 2 (standard of comparison);
- 4.0 wt% pigment sample 3 (transparent, tinctorially strong).
If sample 2 is used as a standard with a hiding power of 100, then the relative opacity of the type with a coarse particle size equals 275, while the transparent variety with a fine particle size has a relative hiding power of 30.
1.8 Areas of Application for Organic Pigments
The considerable increase in demand for coloured pigments over the last three decades is in response to several external factors, primarily of a psychological nature. Advertizing effectiveness and product promotion have become increasingly dependent on the public attention value of coloured materials. On the other hand, both industry and the private sector show an ever increasing tendency to colour products for identification and differentiation purposes.
Organic pigments are used to colour various media. It is useful to distinguish between three primary fields of application: the coatings and paints industry, the printing inks industry and the plastics and fibres industry. In addition, organic pigments are used for special purposes, for instance in office materials and in the mass colouration of paper.
In discussing commercially significant organic pigments and their performance in use, it seems appropriate to define the limitations placed on pigment use by the different fields of application. A given pigment may perform well in one application but poorly in another, and the standards that are acceptable to the pigment user depend on the scope of the product. The chemistry and physics of the medium to be coloured play as significant a role as the processing methods and operational conditions. Ultimately, the choice of pigment and binder system for a specific application almost invariably represents a compromise between optimum technical and bearable economical considerations.
It is useful in this context to focus on the performance of entire pigmented systems rather than to examine the properties of individual components. The properties of a pigment powder, for instance, bear very little resemblance to its ultimate property in use.
As an example, a given pigment, incorporated in one medium, may match grade 8 on the Blue Scale for lightfastness, which is an excellent value; in contrast, a change of application medium may render the same pigment sensitive enough to reach only step 2. Similar observations apply to other technical properties such as weatherfastness, migration fastness and solvent fastness, along with certain colouristic properties, such as tinctorial strength, hue, gloss, gloss retention, and transparency, opacity.
The technical requirements for pigments are subject to constant change because of the development and introduction of new vehicle systems fostered by the proliferation of materials to be coloured. The recurring necessity to replace traditional pigments that are not usable with the newer vehicle systems is a permanent incentive for the development of new products.
However, instead of exploring new pigments, it is often preferable to optimize the properties of one of the already available compounds. This explains why a given type of pigment is often marketed in more than one physical form to provide various colouristic and application properties, each of which is designed for a specific vehicle system or condition of use.
Where this approach fails, new pigments have to be developed. Many patents have been issued in recent years describing compounds to be used as pigments; novel classes of pigments have been proposed. Only very few, however, have entered the market, and even less have turned into a commercial success. This is why R & D laboratories today have shifted their attention towards preferably optimizing the properties of known pigment structures for special purposes. Moreover, notably, economical and environmental concerns remain significant issues in the development of synthetic novelties.
This section lists the primary areas in which organic pigments are employed, along with the respective requirements for pigment use. It is, occasionally, useful to include information as to whether and in which way special demands can be fulfilled.
1.8.1 Printing Inks
The graphics industry is the largest application area for organic pigments worldwide. The portion of organic pigments for printing inks is estimated at approximately 60% of the total market of approximately 240 000 metric tons by quantity.
The portion by value is only around 35%. This reflects that fact that in the printing industry the more economical pigments are mainly used. This is particularly valid for the publication sector, which dominates the printing market with respect to volume. But there are also many high demanding applications such as inks for food packaging, metal decoration, security printing or decorative lamination printing and others.
While for publication inks the emphasis is mainly on colour strength, other colouristic properties and printability, the other applications mentioned above expand the requirements by special fastness properties like light fastness, weather fastness, heat stability, sterilization stability and many more, depending on the designated application.
In past years a clear trend in the printing technology was the acceleration of printing speeds, which also had a deep impact on the formulation of printing inks. This development has now almost ceased. Today the general tendency is more focussed on product safety, protection of the environment and consumer health including also the printing ink industry. Therefore, much attention is directed today to traces of by-products and impurities of organic pigments. This requires pigment producers to improve their manufacturing processes by using suitable raw materials and providing a reliable quality management.
The major printing processes used are lithographic, flexographic and gravure printing. Further important printing processes are screen printing and the modern electronic non-impact printing processes. Each printing process uses differently formulated printing inks and hence often requires differently modified pigments.
The choice of the selected printing process is mostly a question of economic considerations depending on the required printing quality, the number of prints and the adaptation to the latest technical development.
1.8.1.1 Offset Printing
Offset printing belongs to the lithographic printing processes and is, compared with gravure or flexo printing, a quite sophisticated process. The printing plate is flat – image and non-image areas are on the same level. The process depends on physical-chemical properties. The printing areas are lipophilic, whereas the non-image areas of the plate are hydrophilic. This process usually necessitates water or more precisely a fountain solution to prevent ink transfer to the non-image areas. This means that on the printing press the ink comes into contact with fountain solution all the time. The ink must be able to emulsify a certain amount of water, but not too much, and not too little, in order not to create printing defects like tinting or washmarks. Thus, for the manufacturing of offset pigments hydrophilic additives should be avoided. Also important is a thorough washing of the pigment after synthesis as salt residues deteriorate the ink–water balance. The salt content can be measured by means of the specific conductivity and is usually part of the quality control in pigment manufacturing.
In addition, pigments with a high content of water-soluble bivalent metal ions, particularly calcium, are supposed to cause tinting problems as metal ions may deposit on the printing plate. This concerns mainly laked pigments like Pigment Red 57:1.
Components of the ink can also affect the stability of the pH of the fountain solution. For optimum print quality the fountain solution is adjusted to a weakly acidic pH. Through contact of the fountain solution with the ink the buffer may be depleted, causing a shift in pH. This so called pH-drift may be influenced by the pigment, but mostly by impurities or by additives used for the pigment coating.
A very important requirement for offset inks is an excellent flow. Owing to the very thin ink layers produced in offset printing, of 0.7–1.3 µm in four colour process printing, a high pigment loading in the ink is necessary. Without good flow the ink could neither be processed nor could it be properly printed. Today this flow problem is even intensified as mineral oil free formulations for offset inks based on vegetable fatty acid esters are becoming increasingly popular. Diarylide yellow pigments are particularly susceptible to poor flow.
Unfortunately, good flow on high speed printing presses often creates another problem: misting. Usually, good flow of an offset pigment and low tendency to misting is contradictory, as misting increases with lower viscosity. Although this proplem can partly be reduced by modification of the vehicle formulation of the ink, there are specially treated diarylide yellow pigments in the market that show the required rheological properties to overcome this issue.
Offset printing is particularly sensitive to the degree of pigment dispersion. Incomplete breakdown of agglomerates may present problems in the thin layers as produced in offset four colour process printing (ISO 2846-1), affecting the tinctorial strength and the gloss. This occurs with agglomerates that protrude from the surface of the film and scatter much of the incident light. Figures 1.88 and 1.89 show electron micrographs of printed layers. The pigment in this case was dispersed in an offset vehicle at different temperatures, using a three-roll mill (Section 1.6.5.3). The samples for the electron microscopic images in Figure 1.88 were produced with an ultramicrotome, and the images reflect the cross section of printed layers on paper; Figure 1.89 shows the corresponding surface images. The photographs clearly show that increasing the dispersion temperature improves the distribution of pigment throughout the vehicle, produces a smooth surface and thus enhances the gloss.

Figure 1.88 Influence of different dispersing conditions on the degree of dispersion of Pigment Yellow 17 in a sheet offset varnish and on the gloss of prints which were produced from this system.

Figure 1.89 Images of the surfaces of the prints taken with a scanning electron microscope (see Figure 1.88).
A pigment that is not dispersed thoroughly in its medium will also affect the transparency of the resulting printed layer, which is a particularly sensitive issue in multicolour printing. As a result, offset techniques in particular depend on good pigment dispersion. Dispersion equipment, pigment manufacture and resin preparation are designed to cope with this problem. Not only have three-roll mills and other dispersion units been improved continuously but new technologies have been developed, resulting in specialized mechanically agitated ball mills with a high speed shaft and other machinery. Process automation and high throughput make the printing ink manufacture more economical. Modern dispersion techniques have been developed along with the dispersion equipment, presenting a severe challenge to pigment performance. Modern ball mills, for instance, require pigments that are easy to disperse and stable to recrystallization, because even efficient cooling does not entirely protect the printing inks from being exposed to temperatures of 70–90 °C and more during dispersion.
Pigments such as Pigment Yellow 12 and 13 type diarylide yellow pigments, used primarily in offset printing inks, dissolve to a certain extent in mineral oils under these conditions and may therefore recrystallize. This effect is not only dependent on the processing temperature but also on how long the pigment is exposed to the heat. In practice, a pigment may be required to resist such high temperatures for up to several hours. In large-scale production, there is usually some time lag between two production steps. This may cause problems, connected with loss of tinctorial strength and transparency, often to a considerable extent. Moreover, recrystallization also affects the gloss of a product; yellow pigments tend to turn more reddish and become somewhat dull. A printing ink that suffers from extensive pigment recrystallization may perform like an insufficiently dispersed system. Recrystallization also depends very much on the formulation of the ink vehicle. For example, using mineral oils with low content of aromatic hydrocarbons improves recrystallization stability. In addition, improved diarylide yellows are available. Today, mixed coupled diarylide yellows on the basis of Pigment Yellow 13 are often used. With regard to recrystallization, P.Y.176 in general tends to show better behaviour than that of P.Y.174 or P.Y.188 (Figure 1.90).

Figure 1.90 Recrystallization diagram: impact on transparency of the processing temperature.
It is not only the dispersion equipment, however, that defines the dispersibility standards for organic pigments that are targeted for offset application. The composition of the binder also plays a major role. Moreover, permanently improving printing techniques are instrumental in creating severe application conditions. Web offset inks, for instance, have been improved further by reducing the amount of easily wetting alkyd resins and vegetable oils and by increasing the content of hard resins and mineral oil. This shortens the time and decreases the temperature needed to dry the printed layers. Gel varnishes, for example, which are added to printing inks to confer more advantageous printing properties on the host system, wet a pigment with difficulty. Gel varnishes are obtained by reacting varnishes that contain large amounts of hard resin and a small portion of vegetable oil with a metal chelate.
Special-purpose pigments, especially those containing various resins, are manufactured in an attempt to provide systems that are easy to disperse. A disadvantage is the fact that certain resins – especially those contained in the pigment in large concentration – may adversely affect the drying process.
The use of surfactants is not possible, because these otherwise useful agents may disturb the equilibrium between water and ink on the offset printing plate.
1.8.1.1.1 Metal Deco Printing
Metal deco printing can be considered a special area of offset printing. Traditional printing inks, which are processed at 160 °C and more, must be quite thermally stable to retain their colour value during application. Moreover, printed metal sheets that are used in food cans must be capable of being pasteurized or sterilized in the presence of food without degradation. Pasteuriziation is typically carried out at 84 °C in the presence of alkali. The alkali is used to prevent corrosion. Sterilization is typically carried out in water at 120 °C and 2 bar. Some applications require 130 °C and 3 bar pressure or even more. The pigment is usually also expected to be fast to overspraying with clear ‘silver lacquer’ (Section 1.6.2.1). In recent years, however, metal deco inks have been developed that are less demanding in terms of pigment heat stability, for example UV inks.
Often the curing of the metal decoration inks is checked by a ‘rub test’ with methyl ethyl ketone: the pigment must not bleed in that solvent.
Like in normal offset printing, metal deco inks are applied in thicknesses of only about 1 µm, which makes it necessary to use inks containing pigments with high tinctorial strength at high concentrations. Pigments targeted for metal deco printing therefore require excellent rheology as well.
In metal deco applications where the processing temperatures can easily exceed 200 °C, such as for two-piece-cans for beverages, special attention must be paid to the fact that diarylide pigments must not be used due to possible degradation (Section 2.4.1.3).
Sometimes beverage cans require fastness to sweat and saliva (EN 71-3). Laked hydrazone pigments are critical in that regard.
1.8.1.2 Gravure Printing
The main application area of this printing process is used for the printing of magazines, magazine inserts, mail-order catalogues and for the printing of packagings made of plastic film or paper. These two application areas are called publication gravure and packaging gravure printing.
Further important application areas are decorative lamination printing and security printing.
The requirements of the inks in these fields are totally different and subsequently the requirements for the pigments used as well.
1.8.1.2.1 Publication Gravure Printing
Coloured publication gravure printing inks are commonly based on colophony resinates and phenol modified colophony as binder and toluene as solvent. Ready-made printing inks normally contain solvent in excess of 60% and 4–10% pigment. The substrate used is exclusively paper.
Inks (and therefore pigments) designed for use in publication gravure printing must be characterized by extremely good rheology. Printing speeds of up to 60 000 revolutions per minute of the printing cylinder or 16 m s−1 web speed require inks that are able to almost entirely fill a 40 µm deep cell within a fraction of a second. These inks are then transferred immediately onto paper. The performance of the pigment, especially its physical characteristics, largely determine the flow properties of the resulting printing ink. Flow properties are based on very complex phenomena (Section 1.6.8.4) and are far from being entirely understood. The phenomenon of thixotropy in these extremely thin layers, for instance, has not yet been explained on a microbasis. The requirements at different stages of ink application vary and may be contradictory.
Although a structurally viscous or thixotropic printing ink makes it difficult to fill and empty the tiny cells in an engraved cylinder, these are exactly the properties that will make an ink perform well on paper and prevent it from penetrating too deep into the paper. Clearly, the optimum performance is realized if properties like structural viscosity or thixotropy develop immediately after the ink is applied onto paper: the printed dots have sharp edges and the resulting print is clear.
Pigments used for publication gravure inks are also expected to be non-abrasive. A printing ink or ink components that cause mechanical abrasion to the engraved printing cylinder may prematurely damage the printing cylinder, destroying or scratching the cells in the printing surface and thus adversely affecting pictorial reproduction. Some pigments consist of crystals that are hard enough to cause abrasion; typical products that are prone to mechanically attack printing cylinders are often pretested before they are sold and non-abrasive grades labelled accordingly.
Pigments are also selected for their tinctorial strength, which must be very high. Weaker types clearly have the disadvantage of requiring relatively high pigment concentrations to afford equal colour value at a given size of the tiny cells in the printing cylinder. The result of using a tinctorially less strong pigment would be a highly viscous ink that behaves poorly in print due to the higher pigment loading needed.
The organic pigments of interest in publication gravure printing are Pigment Red 57:1, Pigment Blue 15:3 and Pigment Yellow 12. Using these pigment classes inks can be produced that fulfil the requirements for process printing according to ISO 2846-3. In the case of yellow, special amine treated P.Y.12 types have been established.
Although Pigment Yellow 12 types show less of a tendency to recrystallize in toluene, this effect is still quite pronounced. Using pigments with relatively high specific surface areas at the high concentrations that are required to afford a high colour value produces very viscous printing inks. To eliminate these problems, diarylide yellow pigments are used almost exclusively in amine-treated form (Section 2.4.1.4). The resulting printing inks are not only much less viscous, but the pigments are also easily dispersible and exhibit high tinctorial strength. Figure 1.91 shows the shear stress–shear rate curve for amine-treated versus traditional types of Pigment Yellow 12 at equal concentrations in toluene-based publication gravure inks. Amine treatment, which leads to excellent rheology (Figure 1.91) and high tinctorial strength, has made it possible to develop yellow pigment types that satisfy the specifications of technologically advanced printing techniques, including modern high-speed printing.

Figure 1.91 Rheograms of publication gravure printing inks containing 15% pigment. Curve 1: Pigment Yellow 12, Curve 2: Pigment Yellow 12, amine treated.
However, amine-treated pigments also have their problems. The treatment process produces dark red compounds that are easily soluble in toluene and appreciably enhance the tinctorial strength of the resulting printing ink (Section 2.4.1.4). As the ink is printed on to the gravure paper, the dissolved colourant, together with the solvent, penetrates the paper. An ink that does not stay on the surface of the substrate will lose some of its optical effectivity. Furthermore, if the ink penetrates through the sheet of paper it produces a stain on the back of the sheet (strike through). An ink that penetrates through the paper is particularly inconvenient since users frequently employ low cost, thin and light papers in an effort to prevent as far as possible increasing postal expenses in many countries. The demand for less penetrating types of diarylide yellow pigments is satisfied by commercially available special amine-treated pigments, which presently dominate the European market.
The amine treatment of ruby red and blue pigments, which are used in four colour printing Pigment Red 57:1 and Pigment Blue 15:3 type pigments, do not match the effect of the corresponding Pigment Yellow 12 pigments. Amine-treated red and blue pigments are therefore of no commercial value. Besides, these pigments are not as problematic in the non-amine treated form as the amine free diarylide yellow pigments.
1.8.1.2.2 Packaging Gravure Printing
In contrast to publication printing inks, the formulations for packaging gravure inks are much more manifold since there are many different goods to be packed and subsequently many different substrates are being used. Therefore, pigments must be selected with regard to required fastness properties and to the varnish composition used. The solvents incorporated play a large role for the pigment performance and compatibility in an ink.
The list of common solvents ranges from alcohols to esters to ketones, such as methyl ethyl ketone and methyl isobutyl ketone. It is very common to employ a blend of two or more of these solvents to formulate printing inks. Pigments that are used in such inks must be fast to the solvents that are part of the ink system. Furthermore, the pigments must be compatible with the solvents used and form a stable dispersion. While for example pigments in nitrocellulose ink formulations, mainly based on alcohols as solvent, are appropriate they often show flocculation issues in ester-rich formulations.
Moreover, plasticizers, which are used in substrates such as Soft-PVC and which are components of printing inks like nitrocellulose inks, present a challenge to pigment stability. A pigment that satisfies these requirements will show less of a tendency to migrate or recrystallize while the ink is produced, stored and shipped. Recrystallization is a particular problem in inks that are formulated to provide transparency in prints on aluminium film, for example.
Pigments are usually selected for complete fastness to bleeding in an effort to prevent successive printed layers from interacting with each other – a measure that is especially crucial if a white layer is printed last. The problem receives an added degree of complexity in intermediate layers of composite films, which commonly contain pigments that are not sufficiently fast to the respective laminating adhesive (Section 1.6.2.3). Pigments that are to be printed onto packaging films must be particularly fast to various chemical agents and physical influences. Standardized tests have been developed to examine prints for fastness to acid, sodium hydroxide solution, soap (Section 1.6.2.2), detergents, cheese, fats, paraffin, wax and spices (ISO 2836). The pigment is selected with respect to its intended use (Section 1.6.2.3).
To facilitate the printing process, pigments that are targeted for use in packaging gravure inks should also show excellent flow behaviour. The flow properties of a pigmented system are largely determined by the pigment itself; it is a question not only of the particle size but also of the agents that are being used to prepare the surface of the pigment particles. The advantage of optimizing particle sizes to provide good rheology is frequently compromised by simultaneously deteriorating colouristic properties and performance in application. Such changes may sometimes be prevented by suitable pigment preparation or by predispersing a pigment in nitrocellulose resins (NC chips). Nevertheless, it is advisable to always consider a pigment in the context of binder and solvent.
1.8.1.3 Solvent-Based Flexographic Packaging Printing
Another printing process for the printing of flexible packagings is flexographic printing. This process competes with packaging gravure printing. Today, in Europe flexographic printing has a share of about 50% in this application area, a share that is still increasing.
The printing ink formulations are similar to those of the packaging gravure inks, with few differences, mainly with regard to the solvent composition. The drying speed of these inks is slower as they contain retarding solvents such as ethoxypropanol. This solvent is often the reason for additional compatibility issues with the pigment such as flocculation and poor rheology.
Pigments used for inks of flexible packagings often must work in gravure and in flexo printing inks as both are manufactured starting with the same basis.
1.8.1.3.1 Decorative Lamination Printing
An important application area for decorative lamination printing is wood imitations for kitchen surfaces and for floor coverings. The most important requirement is an excellent lightfastness, which typically should be better than 6 compared to the blue wool scale. However, the special processing steps of the laminates require further fastness properties.
The conditions to be met in decorative lamination printing are particularly demanding. Decorative papers for gravure printing are used to produce laminated plastic sheets. Pigment selection is a matter not only of the type of resin (melamine or polyester) but also a function of the processing method.
Polyester sheets require pigments that are fast to certain solvents. A pigment that is used to colour polyester dissolved in styrene must therefore be completely fast to both polyester and styrene. To tolerate the diallylphthalate process, which involves dissolving polyester in acetone, a pigment should be completely fast to acetone.
Pigments that are intended for use in inks for melamine resin sheets are chosen primarily with respect to the particular process characteristics. The list of possible methods includes (i) the high-pressure process with or without an overlay paper or with chipboard coating and (ii) the short-phase technique, which also makes it possible to manufacture polymer films. Working in the absence of overlay paper, that is, without covering the printed decorative paper with a transparent resin-impregnated paper, makes it necessary for a pigment not to show plate-out. Plate-out (Section 1.6.4.1) refers to the deposition of pigment on the impression cylinder or plate, which in turn affects the surface structure of the following articles. The complex nature of this phenomenon makes it necessary to select pigments on an empirical basis only. Fastness to binders and to the solvents that are typically contained in inks to be printed on decorative paper is less of a concern, as experience shows that useful pigments can also be expected to be compatible with the printing ink. When considering the final product, however, excellent lightfastness is a priority.
1.8.1.3.2 Multi-Colour Printing
Multi-colour printing is not a separate printing process, but a method to reproduce natural coloured images. It can be done with any printing process. There are different methods in use, but by far the most important method is four-colour process printing. For this method a natural coloured photograph is separated into four single colour separations by means of special colour filters. The four colours are called cyan, magenta, yellow and black, for increasing the contrast of the print.
Pigments that are designed for process printing have to satisfy special colouristic requirements, which are defined by standardized colour scales. The first scales established fixed the colours for letterpress and offset printing. These standards developed from national to regional standards, like the European Scale for Offset Printing. In the recent years national standardization bodies have agreed on an international standardization. The requirements for the process colours are now fixed in several parts of ISO 2846 and have been extended to other relevant printing methods, as there are sheetfed and heatset offset printing, coldset web offset printing, publication gravure printing, screen printing and flexographic printing. This standard does not specify certain pigments, but determines the colourimetric data and the limits for transparency for each process colour. This is reasonable as the pigments can still be chosen depending on the required fastness properties of the ink and economic considerations.
The standard yellow of ISO 2846 is accessible through a range of hydrazone pigments, such as Pigment Yellow 13, 126, 127, 188, 174 or 176. Quite a number of yellow pigments, whose shades are outside of the tolerance limits, may be adjusted colouristically by appropriate shading. The ultimate shade of a pigment in application is largely defined by the pigment concentration in the ink and by the degree of pigment dispersion. Standard magenta may be approached, for instance, by Pigment Red 57:1, P.R.184 or P.R.185 type hydrazone pigments; standard cyan is produced by using the β-modification of phthalocyanine blue (Pigment Blue 15:3 and 15:4).
However, not all pigments that satisfy the colouristic standards are eligible for use in multicolour printing. The requirements of different areas of application present a considerable challenge to pigmented materials in terms of fastness to overvarnishing and calandering, lightfastness, thermostability, migration fastness, transparency/opacity and rheology. The number of applicable pigments is also considerably restricted by economic considerations, which is especially true for yellow pigments.
1.8.1.3.3 Ultraviolet-Cured Printing Inks
The UV technology is predominantly used in offset, flexographic and screen printing. The application field ranges from labels, premium packagings and metal decoration to a lot of different special applications.
The demand for such inks, which harden through exposure to UV radiation, varies considerably according to the region.
From the pigments point of view the main issues in this application area have been rheology and storage stability with regard to gelling.
At typical concentrations, organic pigments do not significantly affect the drying and hardening of these printing inks. Since UV light causes scattering and absorption by the pigment, some effect is to be expected, but there are no exceptional specifications for pigments that are targeted for this area of application.
1.8.1.4 Non-impact Printing
1.8.1.4.1 Inkjet Printing
Inkjet printing and laser printing are non-impact printing (NIP) technologies.
Inks for inkjet printing in standard office printers are generally coloured with dyes. Pigments are used for higher demands, especially on the light fastness. The development of pigmented inks for inkjet printing is demanding. The particles must be small enough to pass through the very fine nozzles of the printer without causing blackages. Hence the pigments must be dispersed very finely. The dispersions are not allowed to contain particles, aggregates or agglomerates larger than 1 µm. In addition, the dispersion has to be stable and non-flocculating over months or years, even at elevated temperatures. For this purpose pigments such as P.Y.155 (dihydrazone) or P.Y.180 (benzimidazolone) are used for yellow shades, P.R.122 (quinacridone) or P.R.184 (naphthol hydrazone) for magenta, and P.B.15:3 (phthalocyanine) for cyan shades.
1.8.1.4.2 Laser Printing
Laser printers, colour copy machines and most fax machines work by the electrophotographical method. A rotating drum, covered by a photoconducting layer, is electrostatically charged by a high voltage. A laser writes a latent image into the photoconducting layer. Toner particles are transferred to those parts of the image that should be coloured. Subsequently, the toner particles are transferred to the paper. Finally, the particles are fixed on the paper by heating.
The toner particles typically contain a resin as the binder (approx. 90%), pigments, charge-control agents and other additives. The toners are manufactured by an extrusion process followed by milling and classification to obtain particles of controlled sizes, which is typically 6–8 µm. Recent developments aim to produce smaller particles by chemical processes, for example, by polymerization.
Pigments used for laser printing and other electrophotographical processes must satisfy colouristic demands such as hue, tinctorial strength and transparency. Furthermore, the pigments should meet the requirements for their triboelectric properties. The pigment properties depend on particle size, morphology, polymorphic form, impurities and so on. Today P.Y.155 (dihydrazone) or P.Y.180 (benzimidazolone) are used for yellow shades. Magenta shades are obtained by various quinacridone (including solid solutions) or naphtholone pigments or mixtures of them. Copper phthalocyanine is used for cyan shades.
1.8.1.5 Security Printing
Security printing does not denote a particular printing method but an application sector in which one or several well-known printing methods are used, such as offset, letterpress and gravure printing.
Security printing includes printing banknotes, postage stamps, passports, cheques, share certificates and official documents. Tax seals for alcohol bottles and cigarette packs also come into this category. A security print must withstand all physical and chemical stresses for many years or even decades and be unmistakably identifiable at any time. Security prints have to meet the very highest requirements in relation to counterfeit deterrence. This is achieved by a combination of printing method, paper (watermarked papers), printing ink and special elements such as holograms and security threads.
1.8.1.5.1 Banknote Printing
For security reasons, banknotes may only be printed by so-called ‘government printers’ and state-licensed printers. The generally multi-coloured background is printed by various methods such as offset litho, indirect letterpress (letterset) and offset gravure (gravure via a rubber cylinder). Using special guilloche screens, wavy or curved line patterns are produced, which are an important security feature.
In intaglio printing, a tactile ink marking is applied to the substrate by using a viscous ink, a deeply engraved printing plate and very high pressure in transferring the ink onto the paper. With this type of marking, it is possible for blind people to feel the denomination of a banknote by touch.
The oxidation drying printing inks have to meet high chemical and physical requirements. Besides resistance to various solvents, detergents and wash liquors, they also need to have high light fastness, rub resistance and crease resistance.
To obtain the necessary fastness properties, high-quality pigments are generally used for security printing. The range of suitable products covers dihydrazone, dihydrazone condensation, benzimidazolone, naphthol AS, isoindolin, perinone, anthanthrone, dioxazine and quinacridone pigments.
1.8.2 Coatings
The requirements for pigments that are targeted for use in paints depend primarily on the final coated product, but also take into account the paint composition and colour as well as manufacture and process characteristics. In discussing these requirements, a classification system is used based on pigment related characteristics and the drying process. Systems that do not pose a particular challenge to the fastness of a pigment are therefore of no concern in this context. This is also true for most physically drying paints containing nitro and nitro-combination lacquers.
1.8.2.1 Oxidatively Drying Paints
Heading the list are air-drying medium and long-oil alkyd resin systems, which command a good share of the market. They are broad in scope; areas of application range from architectural paints to industrial paints, such as special-purpose automotive repair finishes. These systems contain solvents such as aliphatic (white spirit) and aromatic hydrocarbons, turpentine and higher alcohols, in which most organic pigments are almost insoluble under common processing conditions. Consequently, there is no limit to the choice of pigment with respect to solubility. Lightfastness and weatherfastness are the primary criteria for pigment selection besides colouristic parameters.
1.8.2.2 Oven Drying Systems
Oven drying systems often require highly specialized equipment and production units for application and drying. Such systems are mainly processed in industrial operations. At the present time, most industrial coatings are oven drying (i.e. heat set) systems.
Besides the solvents found in air-drying paints, most industrial finishes also contain solvents that may be more or less aggressive to organic pigments, such as glycols and glycol ethers, including ethyl and butyl glycol, esters like ethyl and butyl acetate, ketones such as acetone, methyl ethyl ketone, and methyl isobutyl ketone, and, finally, chlorinated hydrocarbons and nitroparaffins.
Conditions to be met in oven drying enamels depend also on the composition of the binder. Paint systems containing melamine-formaldehyde or urea-formaldehyde resins, for instance, harden by polycondensation with other resins, such as epoxy resins, short-oil alkyd or acrylic resins at elevated temperatures. Baking is carried out at between 100 and almost 200 °C and may last from a few minutes to more than an hour. A general trend towards energy conservation has shifted public attention towards binders that require low baking temperatures.
Two-component finishes begin to harden immediately upon mixing the components, which makes it necessary to consider the respective pot life. Components may react at room temperature; commercial scale operations, though, are usually accelerated by working at temperatures up to 120 °C. This is true for polyesters containing hydroxy groups, crosslinked with isocyanate, and acrylic resins designed for automotive refinishes. High temperatures are an equally common curing method throughout the wood coatings and furniture industry, which uses partially pigment-free unsaturated polyester lacquers and acid hardening alkyd/melamine or urea systems, respectively.
In recent decades, considerable efforts have been made to restrict or completely eliminate solvent emission during oven drying. Intensive research has focused on developing paint systems that contain very little, or no solvent [239]. Various approaches are possible; systems that have found their way to practical use tend to present a special challenge in terms of pigment selection:
- Materials known as high-solids and medium-solids systems have a higher solids content, and a consequently lower amount of solvent in application, compared with conventional baked enamels [240]. The binders typically used in these systems have a lower molecular weight and are somewhat less efficient wetting agents, but they present no particular problems with regard to pigment dispersion and application. Some years ago effective additives became available that improve the flocculation stability of a pigment in such media. Respective additives, for instance, were developed for dioxazin violet (P.V.23) and quinacridone pigments. The chemical structure of these additives may resemble the chemical constitution of the pigment to be treated. Suitable substances can be added to the system by the pigment manufacturer during the dispersion process or during the production process of the pigment. Special pigment grades are on the market.
- Nonaqueous-dispersion (NAD) systems consist of a blend of dissolved and dispersed binders. The higher solid content of these resin systems – and the correspondingly smaller solvent content – is achieved by partially dispersing the binder. This is of some consequence to pigment dispersion, which is usually carried out in dissolved resin. Where the resin content is not high enough to sufficiently wet the surface of the pigment particles and to stabilize the dispersion, flocculation may occur, especially in pigments with a high specific surface area and at a high pigment concentration. These effects may be avoided or reduced by methods such as increasing the dissolved resin content in the NAD system. In the USA, high solids and NAD systems are currently the systems primarily used for original automotive (O.E.M.) and refinish coats.
- Water reducible systems are usually waterborne, but also contain some portion of organic solvents, for instance, certain types of glycols. The high surface tension of water has a considerable effect on the wettability of a pigment. Highly polar pigments are generally wetted easily by aqueous media and thus disperse readily. Pigments that lack polarity, on the other hand, may be difficult to disperse. By adding suitable additives it is possible to achieve significant improvements. In general, most organic pigments show better dispersibility in waterborne systems, which can be noticed by higher colour strength and chroma compared with solvent-borne systems. Water reducible systems have been discussed extensively in the literature, and a large number of patents have been issued [239, 242–244].
Waterborne systems used for O.E.M. are mainly two-coat systems, that is, a waterborne basecoat with a clearcoat applied atop. In addition to highly improving the gloss, the clearcoat also contains generally light-stabilizer systems and serves to improve the durability of the whole system. Depending on the car manufacturer, various clearcoat systems are in use, such as thermosetting acrylic or two-pack acrylic systems, waterborne systems, acrylic powder coating or powder slurry systems.
Apart from the O.E.M. sector water reducible systems are increasingly in importance with high quality industrial paints and refinish systems as well.
Owing to the very stringent EU regulations on VOC (volatile organic compound) emissions [245], European countries are leading the way with respect to water reducible systems.
Based on the O.E.M. sector the following figures give a picture of the current situation:
| Country | Percentage of water reducible basecoat systems for O.E.M. (%) |
| Germany | approx. 90 |
| Europe | approx. 70 |
| Nafta | approx. 60 |
| Japan | approx. 40 |
- At present, systems cured by ultraviolet light or electron beams [246–248] are used primarily in wood coatings, which contain very little or no pigment; they are employed to a lesser extent in paper coating and other applications. In contrast to the printing field, the comparatively thick layers in which paint systems are typically applied pose somewhat of a challenge to the drying process, especially since pigments absorb UV radiation. Regarding drying properties new results on paint systems revealed that transparent pigment types are more favourably being applied than opaque pigment versions.
The requirements for a pigment used in a powder coating [249, 250] are extremely stringent for various reasons. The shade of a coating, for instance, frequently changes as the material is cured; the effect may be considerable, depending on the temperature and the time [241]. Problems may be avoided or reduced by suitably adjusting the curing agent and the pigment. The colour change has in some cases been shown to result from a reaction between pigment and hardener (Section 1.6.7).
Plate-out is another phenomena that can affect colour change in a powder coating (Section 1.6.4.1). Plate-out is largely due to insufficient wetting of the surfaces of solid particles within the coating. It is possible to reduce or to avoid this effect by selecting suitable pigments and/or by using certain hardeners that are only effective at elevated temperatures.
Pigments that are designed to colour paints are selected partly on the basis of their properties and performance with respect to the applied coating; this includes lightfastness, weatherfastness and fastness to certain chemicals. On the other hand, pigments must also tolerate paint synthesis and processing as well as application of the pigmented medium. The rheology of a system is one of the primary issues, since good flow behaviour facilitates dispersion and application. The list also includes fastness to the respective solvents and resulting properties, such as fastness to overspraying, tendency to bloom and recrystallization stability. Solubility related fastnesses are controlled by the particle size distribution and concentration of the pigment and mainly by the processing temperature (Section 1.6.3.2). The composition of the paint, the choice of solvents and the duration of heat exposure are also important.
These requirements considerably restrict the selection of pigments for oven drying systems. The complex nature of the problem makes it necessary to submit any pigment that is basically applicable to a pilot experiment under the exact processing conditions.
The worldwide trend towards lead-free finishes has given rise to special requirements for organic pigments in this area.
This trend derives mainly from the following reason:
- Preparations containing lead chromate are required to be classified and labelled according to EU Commission Regulation (EC) No 790/2009 (Annex I) as ‘May cause cancer’ and ‘Suspected of damaging fertility’. In practice this is almost causing a prohibition of lead chromate.
In the past additional technical deficiencies sometimes prohibited the use of lead chromate pigments:
- Lead chromate pigments are not sufficiently fast to high sulfur dioxide concentrations.
- Lead chromate pigments are affected by materials containing sulfuric acid, such as soot or moist dead leaves, to which automotive finishes are frequently exposed.
Chrome Yellow and Molybdate Orange Red pigments may be replaced by other pigments to cover the spectral range from greenish yellow to bluish red. Such pigments must be opaque enough to completely hide the substrate in the 40 µm thick layers in which automotive finishes are typically applied. It is therefore necessary for suitable organic pigments to provide considerable hiding power. With several organic pigments, the particle size may be optimized (Section 1.7.3) and the pigment concentration in the lacquer simultaneously increased to satisfy the opacity specifications. The very small specific surface areas of such pigments compared to conventional types make it possible to appreciably increase the pigment concentration without affecting the flow properties, which are vital to ensure easy processing (Section 1.7.8). However, products with an optimized particle size, that is, organic pigments with high hiding power, rarely reach the hiding power of corresponding Chrome Yellow or Molybdate Orange/Red full shade pigments, even if the pigment concentration in the paint is high [251]. This is principally a result of the higher refractive indices of the inorganic pigments (Section 1.7.3) compared to their organic counterparts.
Commonly, blends of organic pigments with suitable inorganic pigments are used, such as nickel titanium yellow, chrome titanium yellow, bismuth–molybdenum–vanadium–oxide or iron oxide.
Formulations that do not contain lead chromate are becoming increasingly important, not only for automotive finishes, but also for architectural paints and other areas of paint application.
The application conditions for pigments in metallic and effect finishes regarding hiding power and lustre, on the other hand, vary considerably (Section 1.7.3). Pigments targeted for use in such systems must be highly transparent. The base coat of a typical two-coat metallic or effect finish usually contains either a water reducible base coat system, mainly in Europe, or a solvent born high solid base coat, especially in the NAFTA (North America Free Trade Agreement) region. The clear top coat commonly consists of acrylic resin and melamine resin, frequently containing UV absorbants to improve the lightfastness and weatherfastness of the entire system.
Depending on the composition of the mill base and the dispersion and application conditions, the dispersion of an organic pigment in the base coat frequently affords colouristially different coatings. Good dispersion is essential to produce the desired high transparency; intensive shear, however, is required to realize this end with pigments with typically fine particle sizes. Aluminium pastes that are dispersed in a binder solution (for metallic finishes), on the other hand, are adversely affected by high shear, because shear changes the structure of the aluminium flakes. Even as the aluminium pastes are manufactured, the particle size distribution is kept as narrow as possible; the smallest particles, which create a grey, dull visual impression, are removed. Applying intensive shear as the paste is incorporated into the base coat seriously compromises the colouristic advantage of fine grain separation. The same applies to the use of pearlescent and other effect pigments.
Notably, however, the dispersion process is of general importance for the application of a pigment in industrial paints. Mill base formulation, various additives, dispersion unit, and dispersion conditions, type of application and operating conditions, and several other parameters (Section 1.6.5) determine the degree of pigment dispersion in the finished paint. Neglecting the cause–effect relationships and interactions will disturb further processing, an effect that is easily revealed in laboratory experiments. Rub-out phenomena (Section 1.7.5) are among the more common of these effects.
A spraying–pouring test has been devised to facilitate comparisons between application conditions. Films made of the same paint, applied by spraying or pouring, are compared with respect to the gloss in full shade and the tinctorial strength of the reduction. It is possible to try and eliminate differences by adjusting the mill base formulation and the dispersion conditions. Specialized additives for paints are available for systems that do not respond to these attempts. These agents are very useful in avoiding or eliminating floating in pigment blends. The theoretical background of floating and other reasons deployed to explain rub-out effects are discussed in the literature [252–254].
A wide variety of dispersion equipment is employed by the paint industry to produce oven drying paints. All have advantages and disadvantages for pigment application, which have to be weighted in each case. A unit for a particular purpose is selected on the basis of the tendency of the pigment to recrystallize and the degree of pigment dispersion at optimized mill base formulation. Equally important are energy considerations and product output, that is, the amount of paint produced in a certain time. The list also includes the type of operation, that is, by continuous or batch process, and cleaning a unit before colour change. Importantly, not all types of mills are suitable for pigments that are difficult to disperse.
Several speciality areas pose a particular challenge to suitable candidates for pigmentation. One example is the long-term heat stability in speciality paint systems for radiators. Several hydrazone and polycyclic pigments have been tested for colour stability by exposing the coated metal sheets to 180 °C for 1000 h and subsequent irradiation in an accelerated weathering cabinet. Compared to unheated specimens, the lightfastness of the thermally treated samples is inferior by an average of one step on the Blue Scale.
Another speciality area is coil coating, which involves coating metal coils by continuous operation. Modern roller systems afford speeds of up to 200 m min−1. Most coils are made of cold-rolled and surface treated steel, aluminium or alloys of the latter with manganese or magnesium. Coating systems are based on alkyd or acrylic resins, oil-free polyester, silicone-modified polyester or acrylic resin, poly(vinylidene fluoride) or poly(vinyl fluoride). Water-reducible systems, mainly based on acrylic resins, have been developed for aluminium as well as for steel coils [255–258]. Drying is carried out by continuous operation in gas- or oil-heated multichamber ovens.
The layers are heated to 280 °C or higher for a few seconds up to several minutes. The coated metal coils are then processed, that is, deformed, profiled or stamped. The resulting coated products are broad in scope; they are employed in various areas from packaging and the vehicle industry to household items and building materials.
The conditions to be met by pigments that are targeted for this area of application vary considerably, depending on the purposes and the coating system. For products that are sufficiently heat resistant it is important to consider the solvent fastness and recrystallization stability of the pigment in relation to the respective coating system, its composition and processing history. Organic pigments rarely satisfy the demand for extreme weatherfastness over a period of several years, which is crucial, for instance, in applications such as sheet metal panels for wall covering. Polycyclic pigments and some very fast hydrazone pigments are commonly selected where the requirements are less stringent. A pigment that adversely affects the mechanical properties of a coating, such as its hardness or scratch fastness or extreme elasticity, is not acceptable in practice.
1.8.2.3 Emulsion Paints
Emulsion paints are based on aqueous synthetic resin dispersions, which afford a lacquer-like paint film. The resin dispersions commonly used by the paint industry contain water as the carrier phase. A large number of such dispersions are available, based on different resins such as poly(vinyl acetate), which may be employed as a copolymer with vinyl chloride, maleic dibutyl ester, ethylene, acrylic acid esters, polyacrylic resin and copolymers of the latter with various monomers, as well as styrene–butadiene or poly(vinyl propionate). These dispersions are highly balanced systems, prepared by adding emulsifiers to the inner or outer dispersion phase.
The dispersions are stabilized by agents such as cellulose derivatives, poly(vinyl alcohol), starch or gelatine, which more or less swell in the presence of water and act as protective colloids. Using pigment powders may give rise to problems, because increasing the solid surface area within the dispersion will almost invariably disrupt the equilibrium between the two phases. Therefore, it is most advantageous to employ pigment preparations that contain surface active agents that do not noticeably affect the dispersion equilibrium. Preventing the viscosity from increasing and coagulating the entire paint system is critical to the success of a system. Moreover, these pigment preparations sometimes lose tinctorial strength as they are stored, owing to wetting, dispersion and stability deficiencies. Less solvent resistant pigments may also tend to recrystallize, a trend frequently enhanced by surface active agents. Loss of tinctorial strength, a phenomenon that is commonly observed in pigmented paints, may arise for several reasons. Inadequate wetting of the particle surfaces will affect the tinctorial strength of a pigmented system, and every care must be taken to ensure adequate wetting in the presence of water-immiscible solvents, large amounts of antifoam or later addition of plasticizers [259].
Depending on the area of application, pigments in such paints must satisfy several additional specifications. Exterior house paints, for instance, should not only exhibit excellent weatherfastness but also tolerate lime and concrete (Section 1.6.2.2).
Apart from pigment preparations that have been developed specifically for aqueous emulsion paints, there are also products known as multipurpose tinting pastes that show outstanding fastness. They have the advantage of being useful not only in emulsion paints but also in solvent containing architectural paints.
Multipurpose tinting pastes usually contain pigment and hydrophilic solvents, sometimes some amount of water, and suitable wetting agents, which define the equilibrium between hydrophilic and lipophilic character. Conditions to be met in each case depend on the type of paste and on the method by which it is produced and also on the intended use of the coating or paint.
1.8.3 Plastics
Owing to the growing number of different plastics on the market organic pigments have to meet increasingly high requirements, of which only heat resistance and dispersibility will be considered here. Within the scope of this book only the most important types of plastics will be discussed.
In making a sensible choice of pigments, besides the plastics the crucial factors are the processing method and the required fastness level.
Since a very wide variety of additives such as antioxidants, UV absorbers, HALSs (hindered amine light stabilizers), plasticizers, slip agents, antislip agents, blowing agents, antistatic agents, flame retardants and fillers are added to virtually all plastics, the self-colour of the plastics used is obviously a key element in the ability to produce the desired colouration. Another example that can be given is the wide variety of stabilization systems for PVC, which have a strong self-colour ranging from white (in the case of lead stabilizers) to orange (in the case of barium–zinc stabilizers) and which also differ greatly in opacity – characteristics that have to be taken into consideration for colouration.
Most plastics like polyolefins and polystyrenes and their derivatives such as ABS (acrylonitrile–butadiene–styrene) and SAN (styrene–acrylonitrile) are supplied by the manufacturers in ready-to-use form with most of the above-mentioned stabilizers or, if required, additional additives are needed to stabilize with, for example, antistatic agents and HALS agents. In contrast, in the case of other materials (e.g. PVC) it is the end user who adds the additives, pigments or preparations. This is normally done on fluid or high-speed mixers, although in the past gravity mixers or tumble mixers were also used. The mixture is then homogenized on mixing rolls, kneaders, planetary extruders or twin-screw kneaders and further processed.
The processing methods are very much governed by the desired end product, for example injection moulding, injection blow moulding, extrusion into blow mouldings, flat film, blown film, profiles, fibres, calendering, foaming and coating. Since the tendency in the plastics industry is towards ever-thinner wall thicknesses for films, fibres and also bottles, standards of dispersibility are being raised all the time and can often only be met by products that comply with increasingly high requirements. With normal processing on injection moudling machines or single-screw extruders, the shear forces necessary for dispersion of the pigments are not applied. This work is normally undertaken by special firms (masterbatch manufacturers). It is carried out on specially equipped twin-screw extruders, kneaders or, for liquid dyes, on triple-roll mills or bead mills in a suitable carrier material. The pigment concentration of preparations depends on the end product and the desired concentration in use.
In pigment preparations the pigment is present in dispersed form, which with appropriate choice of carrier material enables the plastic to be coloured uniformly, homogeneously and reproducible under normal processing conditions.
Dispersibility of the pigments for the plastics industry is tested by the filter test according to EN 13900-5 (Figure 1.92) [260].

Figure 1.92 Determination of the dispersibility of pigments in plastics by the Filter test method according to EN 13900-5.
In this test a specific amount of pigment in prepared form is forced through a fine screen pack by an extruder and the rise of pressure upstream of the screen pack is measured. The calculation is expressed by the formula:
where P1 is the melt pressure of the plastic without pigment and P2 is the melt pressure after the pigment is added. The tolerance in this test is 2 bar g−1 pigment.
Pigment preparations are marketed in solid form as granules and powders or as pastes. They normally contain between 5% and 50% pigment and each is suitable for only a few plastics depending on the type of carrier material used. General-purpose preparations are also available but are only of minor importance compared to the preparations tailored to particular plastics. Moreover, general-purpose preparations require general-purpose pigments, which simply increase the cost of colouration unnecessarily.
To ensure good dispersion of the pigments in a preparation and subsequently good distribution in the end product, it is necessary to increase the flowability of the preparation above that of the plastic being coloured. This is achieved by using a carrier material with a low molecular weight (wax), a copolymer or increased plasticizer content.
In recent years there has been an increasing tendency to use pigment concentrates. These are defined as pigment preparations that contain two or more pigments and produce a defined shade when mixed with a specific amount of the un-coloured plastic. Occasionally, pigment mixtures that have been mixed previously in the correct ratio to give programmed shades are still used for this purpose. These mixtures are usually produced in high-speed mixers.
Organic pigments often differ in their dispersion properties in a plastic. Therefore, mixtures of such pigments can cause considerable difficulties in adjusting the shade, even with slight variations in processing temperature, as a result of the associated different levels of plasticizing.
Plastics are generally marketed and processed in powder or granulated form.
Certain processing methods have proved successful for colouring with pigment or pigment preparations [261]. For example, plastics in powder form are often premixed with powder pigments in fluid mixers. The pigments are then dispersed while the plastic is being processed, for example in the case of PVC in kneaders or mixing rolls or on calendars.
In the case of polyolefins the dispersing performance of single-screw extruders and injection moulding machines is not sufficient to achieve high-quality colouration (fibres, film, tape) with powder pigments or pigment mixtures.
For colouring plastic granules, granular pigment preparations are primarily used, as well as paste preparations, for example pigment–plasticizer pastes. Granular pigment preparations bring advantages in automatic metering by volumetric or gravimetric metering units and metering pumps.
They are, however, of only limited suitability for colouring plastics in powder form. Their distribution in the plastic is often inadequate and the risk of de-mixing exists particularly with pneumatic conveying.
The solvent fastness and insolubility of the pigment in the plastic and all its constituents, particularly under the processing conditions, are often of fundamental importance when selecting suitable pigments. Thus, the problems of blooming and bleeding covered by the term migration are based on the complete or partial ‘dissolving’ of the pigment in the plastic at its processing temperature.
Recrystallization too is attributable to the pigment having a specific solubility in the plastic. As in other media, it is demonstrated primarily in a change of transparency or opacity in transparent colourations and in the depth of shade in white reductions. Lack of recrystallization stability becomes evident for example in the manufacture and processing of pigment–plasticizer pastes and in various polymers at elevated processing temperatures.
The use of pigments in several plastics is restricted by high processing temperatures. An important factor here is the residence time, that is, the duration of thermal stress. For this reason, if coloured regrind is added, its previous exposure to high temperatures must be taken into consideration. Furthermore, the pigment concentration as a function of the heat resistance and of the ratio of coloured pigment to white pigment must be taken into account. This should be determined by means of the limit concentration, that is, the ratio of lowest possible amount of coloured pigment to titanium dioxide, in the thermal stability test. The required heat resistance differs from plastic to plastic.
Several organic pigments can cause warping of certain thick-walled, large-area, non-axially symmetrical injection-moulded parts such as bottle crates, where they act as nucleating agents for partially crystalline polymers.
The light fastness requirements of plastic colourations are met by many organic pigments but are similarly dependent on the ratio of coloured pigment to titanium dioxide. The stability of the plastic used should also be taken into consideration here.
The light fastness of a pigment often differs greatly from one polymer to another; the pigment can have long since faded in one plastic after exposure to light, whereas in another it shows no signs of being affected by light. Thus, with plastics, too, the light fastness can always be stated only for the entire pigmented medium. Owing to their strong inherent scatter, lead stabilizers or antimony trioxide added as flame retardants to PVC can influence the light fastness of the plastic system as strongly as titanium dioxide in white reductions. The comparison values given in the special sections of this book always refer to colourations of identical standard depth of shade. These are adjusted according to DIN 53 235/2 [262].
Standards exist for determining the light fastness of coloured pigmented plastics in daylight and in xenon arc light, that is, in accelerated exposure equipment, and for determining the weathering fastness.
With various plastics the compatibility of the pigments with the polymer system being coloured must be borne in mind. In other cases any influence of the pigments on the physical and mechanical properties of the plastics used must be eliminated. These requirements will be considered in greater detail for the plastics concerned.
Pigments for plastic articles, for example films, that are used for packaging food or cosmetics must not be only migration-fast and extraction-resistant but also physiologically safe. Furthermore, several countries have different regulations that contain specific purity requirements.
There are only a few pigments that meet all requirements mentioned here, as well as any additional ones imposed, and which are also particularly cost-effective. In general, higher fastness requirements can be met only by more expensive pigments. As in other fields, in selecting pigments for a specific colouration problem for plastics, a compromise is therefore made between the fastness requirements and the price. In the plastics sector inorganic pigments are included prominently in these considerations. Whether it is better to use organic or inorganic pigments in a particular case depends on technical and economic considerations and regulations in the individual countries.
Organic pigments have to be used if transparent colourations are desired and high colour intensity, especially for thin-walled articles such as films and fibres, is needed. Organic pigments are also usually required for multicolour printing on films. If, on the other hand, high opacity coupled with a very pale shade is required, which additionally has to have high light and weathering fastness, inorganic pigments are preferred.
Often the advantages of inorganic and high-quality organic pigments are combined; the inorganic pigment with the lower tinctorial strength is usually present in excess to achieve the desired opacity, whereas the organic pigment used in smaller amounts produces primarily the colour intensity of the pigment mixture and the brilliance of the shade.
The following section describes the major types of plastic, and the properties and processing conditions that are important for colouration with pigments. It also presents the requirements for pigments used to colour plastics and provides test methods.
1.8.3.1 Polyolefins
Polyolefins are quantitatively the largest group of plastics and their importance for colouration with organic pigments is correspondingly high.
Depending on the starting materials (monomers) and the density (processing temperature), polyolefins may be classified according to the following principal groups:
| LDPE | low-density polyethylene |
| LLDPE | linear low-density polyethylene |
| HDPE | high-density polyethylene |
| PP | polypropylene |
The respective processing temperatures, which determine the criteria for pigment selection, are as follows:
| LDPE | 160–220 °C |
| LLDPE | 220–240 °C |
| HDPE | 190–300 °C |
| PP | 200–300 °C |
At any given temperature, the flow property of a system is defined by the density and the molecular weight of the plastic and characterized by the melt flow index. Being partially crystalline, polyolefins scatter very little light and therefore tend to appear lighter on colouration, depending on the processing temperature. Another type of lightening effect is observed in stretched and foamed polyolefins.
Polyolefins are processed primarily by injection moulding and extrusion techniques.
Ethylene/propylene products reign supreme amongst the copolymers. They are elastomers. Plastics containing about 20% or more propylene perform like natural rubber and can be cured by peroxide crosslinking. They are faster to chemical and to ageing than other types of natural rubber.
Polyolefins are produced in various forms: HDPE and PP are produced as powders, while LDPE emerges from the melt preferably in the form of lenticular granules. All types, however, are supplied primarily as granules. As a rule, any thermoplastic transformation of a polymer powder into a granulate is carried out in the presence of additives. This is also partly true for pigments.
Unsurprisingly, since polyolefins have a low glass transition temperature (Section 1.6.3), pigments that are partially soluble in polyolefins tend to migrate. Like in other media, this trend is concentration and temperature dependent. However, the type of polyolefin, especially its density and molecular weight, influences the tendency of a pigment to migrate in LDPE to a much larger extent than in other plastics. As a rule, pigments migrate more easily in LDPE than in HDPE or in PP. HDPE and PP, for instance, may safely be coloured by organic pigments that migrate in LDPE.
The trend amongst pigments to migrate in LDPE, which includes both bleeding and blooming, increases with increasing melt flow index and decreasing molecular weight of the polymer, respectively. Additives such as lubricants or antistatic agents may also play a role.
Several organic pigments cause distortion in certain types of polyolefins, especially in HDPE. Pigments act as nucleating agents in such partially crystalline plastics; that is, they promote crystallization, which creates stress within the plastic product (Section 1.6.4.3). These pigments also enhance the shrinkage of polyolefins, particularly in the direction of the flow.
The effect of heat ageing on a polyolefin, as on modified polystyrene, is considerably influenced by particular pigment lakes. Heavy metal ions, especially copper, manganese and iron adversely affect the thermostability of polyolefins, while sulfide containing inorganic pigments have a distinctly improving effect. The response of a system to additives or impurities is tested by measuring the loss of tensile strength after exposing the polymer to heat. The polyolefins are stored at temperatures close to the melting point of the crystallites and the pigmented and pigment-free test samples compared for brittleness.
The light fastness of pigments in a polyolefin, as in other polymers, paints or printing inks, is always measured for the entire pigmented system, including the polymer with all its additives. This aspect gained an added degree of importance since it became increasingly important to blend plastics, especially PP and HDPE, with sterically hindered amines, known as HALS stabilizers (hindered amine light stabilizers), so as to protect the plastics material against light and especially weather. However, some caution should be exercised in combining pigments and stabilizers, because the effectivity of these agents is inferior in the presence of certain pigments.
The specific requirements for organic pigments in terms of heat stability result from the temperature levels at which individual polyolefins are processed. Standards have been developed to test the heat stability of pigments in polyolefins [267] (see also Section 1.6.7).
The dispersibility of an organic pigment that is to be incorporated in a polyolefin is of particular importance. This is especially true for the colouration of extrusion films and for HDPE or PP ribbon made from stretched blown film or from sheeted extrusion film, as well as for the coating or melt spin dyeing.
As a result of the high processing temperatures and the consequently high degree of softening, only limited shearing forces are available to disperse pigments in polyolefins. Pigment dispersion is therefore far from adequate. Therefore, pigment preparations, usually in combination with polyolefins as a carrier, are frequently used to colour polyolefins. Not only is the tinctorial strength improved, but the safety of the operations is also superior. Poor dispersion causes filler specks, holes in films and other faults, which are avoided by using pigment preparations. Nowadays there is a considerable trend towards using colour concentrates, which are easy to apply in metered amounts and which facilitate the production of exactly defined shades (Section 1.8.3).
Pigment powders continue to be used in thick-walled articles, such as extruded sheets, rotational moulded articles and in injection-moulded products.
Several mixing and processing techniques have been used successfully to colour granulated or powdered polyolefin with pigments or pigment preparations. Powders, for instance, are processed primarily with high-speed mixers, while granulated types respond better to slower mixers.
Pigment/polyolefin paste preparations containing between 20% and 70% pigment are also used. They are manufactured by means of three-roll mills, agitated ball mills or dissolvers or similar equipment. Such preparations are employed primarily to produce bottles, injection-moulded articles, or extrusion sheets. These preparations have the advantage of being applicable in volumetric doses and may be premixed in slow mixers or gravitation mixers to uniformly adhere to the plastic granulate.
Details are discussed individually in the literature [270].
1.8.3.2 Poly(vinyl chloride) (PVC)
Owing to rising oil prices, PVC is once more becoming increasingly attractive in that it consumes only half as much oil as polyethylene, while the other half consists of rock salt, a virtually inexhaustible raw material. The discussion of PVC from an ecological point of view has become much more objective in recent years. Objections on health grounds to its constituents, for example plasticizers and stabilizers, have been refuted scientifically. Established methods for recycling materials will in future be supplemented by those for raw material recycling.
Vinyl chloride has been known for over a 100 years and its polymerization to poly(vinyl chloride) (PVC) was achieved in 1912. Industrial-scale production of this plastic began in 1927. The worldwide PVC production capacity in 2012 was 54 mio t, the consumption 38 mio t. The estimated global demand in 2020 may reach 50 mio t.
PVC is still the most versatile plastic. One reason for this is the numerous variations made possible by the method of manufacture of the polymer, namely, by copolymerization with other monomers and their processing. Thus, PVC can be thermoformed on all conventional processing machines if the slight thermal damage is taken into consideration. Machining is easy and the material can be bonded, bent, welded, printed and thermoformed.
A distinction is drawn between bulk, suspension and emulsion PVC on the basis of different polymerization methods.
Three methods are used to manufacture PVC. The suspension method is used globally in about 90% of cases; in Western Europe, with its applications based on specialties, the figure is still at least 86%. Besides the high-volume standard grades, high-quality products such as graft polymers and copolymers, pastes and paste extender resins are produced by this method. The emulsion method is used primarily to manufacture special paste making grades. Its share of capacity is 9% in Western Europe, but only 6% globally. The share of the bulk method is even smaller, accounting for 4% in Western Europe and 3% globally. All in all, the bulk method is becoming increasingly insignificant as a technology; no more new capacities using this method are being created.
The dispersibility of the pigment in PVC, as in other plastics, is a property that determines its use. Here, too, the processing parameters, especially rising temperature, are changing for economic reasons and are less and less suitable for achieving a good degree of dispersion. Thus, for example, temperatures up to 200 °C and above are often used in calendering, added to which the haul-off rate of the films is increasing and the rolling times are decreasing.
A method to test the dispersibility (difficulty of dispersion) of pigments in plasticized PVC was standardized in 1975. However, for various reasons, this method of cold rolling on a laboratory rolling mill of specific design has not proved successful in practice. New test conditions and test materials were elaborated in comparative tests involving pigment manufacturers and processors, and the results will form the basis of an improved standard. According to this the colourations will be carried out in a specified PVC compound firstly at 160 °C (low shear forces) and secondly at 130 °C (higher shear forces) under defined conditions.
Note here that exact and reproducible colourations of PVC in the laboratory and in the plant present problems and can be achieved only if specified working conditions are observed and special equipment is used.
In many cases pigment–plasticizer pastes are also used for colouring PVC. Because of the mixing or embrittlement gap in the PVC–plasticizer system, which ranges from 5% to 18% for dioctyl phthalate for example, such pastes have little or no suitability for unplasticized PVC compounds. In this concentration range, which is specific for each plasticizer, no plasticizing effect is achieved – on the contrary the additive causes embrittlement of the PVC.
Organic pigments can be dispersed readily in plasticizers, for example with the aid of triple-roll mills. However, the throughput here is low because of the lack of smoothness of such pastes compared to other systems such as letterpress or offset inks, which is why attrition mills are better.
The pigment content of plasticizer pastes is normally between about 20% and 40%. The addition of special dispersing agents to several pigments in the manufacture of such pastes has brought advantages. The additives promote accelerated wetting of the pigment and enable the pigment content of the pastes to be raised and the dispersing process to be shortened, for example the number of dispersing operations on a triple-roll mill to be reduced. Since some pigments differ considerably in their dispersion properties from others, when using pigment–plasticizer pastes it is highly advisable not to disperse together mixtures of pigments necessary for adjusting the shade. Pastes containing only one pigment should be mixed homogeneously to adjust the shade. Recrystallization of certain pigments can occur in manufacture, storage and processing of the plasticizer pastes. Pigment–plasticizer pastes are the neatest means of colouring PVC spread-coating pastes (PVC plastisols), though powder pigments too can generally be readily processed in these. This is due to the good wetting characteristics of the plasticizer mentioned earlier.
A reduction in the plasticizer content leads to a better degree of dispersion at a given temperature owing to an increase in viscosity of the plasticized plastic, but even in plasticizer-free PVC complete dispersion of the pigment is not achieved on the roll mill under the dispersion conditions chosen.
A pigment preparation of the same pigment with a copolymer of vinyl chloride/vinyl acetate as carrier material shows by contrast that the temperature has only a slight influence on the tinctorial strength. The pigment is already in dispersed form in the preparation and is distributed uniformly in the similarly plasticized plastic when incorporated with heat after plasticizing of the carrier material. Here too, however, differences in tinctorial strength can be recognized colourimetrically, though these are near the tolerance limits of the methods employed. Such preparations are equally suitable for plasticized and unplasticized PVC.
Diluents, mainly volatile aliphatic or aromatic hydrocarbons such as petroleum ether, dodecylbenzene or glycols, are often added to PVC pastes to lower the viscosity. These do not have a gelling effect and are evaporated off before gelation is commenced to prevent fine cracks or bubbles in the coating. Pigments for this application are required to have adequate resistance to the solvents used at the processing temperatures.
PVC spread-coating pastes are normally manufactured in high-speed planetary mixers that can be evacuated. It is best to add the plasticizer first, stir in the pigments or pigment pastes and only then to add the solid components in portions. In pigmenting plasticized PVC compounds, on the other hand, it is necessary to mix the pigments with the PVC before adding the plasticizer. Pigment–plasticizer pastes can, however, be added to the PVC at the same time as the plasticizer.
PVC is processed by all methods suitable for thermoplastics. Consequently, the required heat resistance and migration fastness of the organic pigments differ greatly.
Several standards are available for determining these properties. They relate both to the composition and the manufacture of the basic compounds of unplasticized and plasticized PVC and of PVC plastisols and the production of the individual test specimens as well as to the performance and evaluation of the tests [263–265].
The heat resistance of the pigments in unplasticized and plasticized PVC is determined by the long-term milling test, in which milled sheets treated on laboratory mixing rolls under defined conditions at 190–195 °C for 10, 20 and 30 min are compared colourimetrically or visually against an untreated milled sheet after compression moulding. The heat resistance of pigments in PVC pastes (plastisols) is determined in a heating cabinet. Defined test specimens are kept for 30 min at 180 °C or 8 min at 200 °C and compared colourimetrically or visually against a specimen left at room temperature [266].
A heat resistance of 200 °C for 5 min for organic pigments is often taken as a guide value for practical use in PVC, with good stabilization of the PVC being a pre-requisite. In a high white reduction certain shade, changes caused by inadequate heat resistance do, however, occur in certain cases under these conditions.
Substantially higher standards of heat resistance are required for certain processing variants. One such case occurs with foam floor coverings and wall coverings based on PVC manufactured in a certain way. Here, the PVC spread coating paste applied to a substrate such as jute, felt or paper is first printed by gravure printing and then covered with a transparent PVC layer. The PVC layer contains a blowing agent, generally azodicarbonamide, which decomposes in the presence of zinc oxide on gelation and expands the PVC layer. Temperatures of about 150–210 °C are used for pre-gelation, while those of 220 °C and above are used for actual gelation, for about 5 min in both cases. Foam formation can be controlled and a structured surface achieved by using inhibitors, for example certain acids or benzotriazole, which may migrate from the printed layer into the PVC layer. Pigments for this use must naturally have the necessary heat resistance. Diarylide pigments, for example Pigment Yellow 83, decompose at processing temperatures above 200 °C to form monoazo dyes and other cleavage products and are unsuitable for such applications.
Detailed methods are also specified in various countries for determining the fastness to bleeding [267]. Plate-out frequently occurs in the colouring and processing of PVC particularly on calenders and roll mills.
An effect similar in appearance to plate-out that affects PVC in particular is chalking.
Pigments for articles that are permanently exposed to the elements, such as garden furniture, facade claddings or roller shutter profiles, must have high weathering fastness. Only a few organic pigments have adequate fastness to long-term weathering. The method used to stabilize the PVC is an important factor here.
In pigmenting the wide variety of PVC grades it is assumed that the pigments are compatible with the polymer and all its additives and do not react chemically, which is generally true of organic pigments. One exception is laked pigments, some of which form the metal-free pigments in the emulsifier-containing emulsion PVC as a result of hydrolysis. Such a process is accompanied in most cases by a change in shade and fastness properties.
Azomethine metal complex pigments replace the metal with tin stabilizers, resulting in a change in shade. In the case of manganese-laked pigments trouble can also be expected in the presence of epoxy compounds. Pigment preparations based on epoxidized soya bean oil are normally used instead of diisodecyl phthalate pastes in the automotive sector, for example for colouring PVC roofs.
Similarly, one frequent prerequisite is that the pigments used have little or no effect on the physical and mechanical properties of the plastic. One example here is the change in rheological properties of PVC plastisols or of PVC melts during processing.
Another example is the influence of the electrical resistance of PVC cable insulation. This is caused not by the organic pigment itself but by ethoxylated surfactants, which are added as auxiliaries in the manufacture of these pigments, especially hydrazone pigments. Contrary to a repeatedly expressed view, a possible electrolyte content, which laked hydrazone pigments for example can have, has no effect on the dielectric properties of PVC [268].1
Some pigment manufacturers offer special product ranges with verified dielectric properties for this purpose.
Pigment preparations are frequently used in the PVC cable industry for colouration. The pigment content is often selected in such a way that one part by weight of the preparation is used to colour 100 parts by weight of the polymer compound. The shades and colour codes for cables and insulated lines are specified in standards in various European countries [269].
As far as the trend in the individual market segments is concerned, unplasticized PVC is the clear winner with a good two-thirds of all applications (Table 1.8). The sole exception here is the bottle segment, in which substitution by PET has forged further ahead. Pipes and fittings are still the largest single application, followed by profiles and rigid films, which display steady growth. The profile segment profits primarily from the PVC window market, which is steadily gaining ground and has already achieved a market share of 55% of the total window market in Germany. The plasticized PVC sector is characterized, on the one hand, by stagnating/shrinking segments (cables, plasticized films) while, on the other hand, floorings and technical coatings have recorded positive growth rates in recent years.
Table 1.8 Consumption breakdown for PVC by fields of application in Western Europe.
| Applications | Share (%) |
| Unplasticized PVC | 67 |
| pipes and fittings | 26 |
| profiles | 23 |
| films | 11 |
| miscellaneous | 7 |
| Plasticized PVC | 33 |
| films | 7 |
| cables | 8 |
| floorings | 5 |
| coatings | 4 |
| miscellaneous | 9 |
| Total | 100 |
PVC production capacity and production worldwide had grown to 48 million tons per year in 2009. The highest PVC production is in Asia and Europe with more than a half of Asian PVC produced in China.
1.8.3.3 Polyurethane
Polyurethane (PUR) is one of the most versatile plastics, due to the wide variation of the starting materials and by the use of practically all processing methods known in the plastics sector. Basically, what has been said in relation to the plastics considered so far can be applied to colouring PUR.
Thus, virtually the same organic pigments are recommended for colouring thermoplastic PUR as for plasticized PVC. Pigment migration processes are equally as important in both cases. This is particularly so in colouring PUR leather cloth, for which a high pigment concentration is normally used.
Like PVC, PUR can be processed together with plasticizers. However, thermoplastic PUR grades for plasticizing PVC are also marketed for use in combination with plasticizers or on their own. Their advantages lie in improved oil and abrasion resistance and in preventing exudation of plasticizers.
Pigment preparations are also marketed for colouring thermoplastic PUR. Their carrier materials range from vinyl chloride or vinyl acetate copolymers, such as are used for PVC, through low-molecular polyethylene to PUR itself.
Coating of textiles with two- and one-component PUR has achieved major importance in the manufacture of leather cloth. For the latter, a distinction is drawn on the basis of the solvent, that is, between aromatic (solvent: dimethylformamide and tetrahydrofuran with the addition of methyl ethyl ketone, toluene, etc.) and aliphatic grades (solvent: often isopropanol–toluene mixtures). This requires the organic pigments used for colouring to be largely insoluble in the solvents concerned. Good solvent fastness, particularly to dimethylformamide, is often not a characteristic of organic pigments.
Laked hydrazone pigments are completely unsuitable for this purpose because they may also lead to bleeding in wet conditions.
The powder pigments are dispersed in one part of the PUR solution to be coloured on ball mills, sand mills and other enclosed dispersing equipment with a pigment content of about 20–40%. The pigment pastes are then mixed homogeneously with the PUR solution; during this operation flocculation must be prevented. Pigment preparations can also be used, but it must be ensured that their carrier material is soluble in the solvent system in question and that fastness properties during use and handling of the artificial leather cloth are not adversely affected.
PUR foams – increasingly integral foamed parts for vehicles and the furniture industry in particular – are also important as far as colouring with organic pigments is concerned. The required heat resistance of the pigments for this sector is in some cases very high. For PUR foaming by the high-pressure method, in which isocyanate and polyol are sprayed through narrow nozzles under high pressure, it is necessary for the pigments used to be fully dispersed to prevent production problems due to blocked nozzles. Normally, pigment preparations are used here, too. Their carrier material is in most cases either involved in the reaction of the isocyanate in PUR formation or it takes part in foam formation. Further details of the carrier material of these preparations, for example its OH number, are therefore provided by the manufacturer.
1.8.3.4 Polyamide, Polycarbonate, Polyester, Polyoxymethylene
Of the large number of other plastics on the market only polyamide (PA), polycarbonate (PC), polyethylene terephthalate (PETP) and polyoxymethylene (POM) maintain an important position. Pigmentation of these polymers largely follows the rules described above. In the case of polyamide for injection moudling and extrusion the choice of high-temperature-resistant pigments is additionally narrowed by the weakly alkaline and reducing character of the polymer melt. For this reason, preliminary testing of organic pigments in the polyamide grade used should be carried out.
The range of organic pigments available for colouring the different plastics varies according to the requirements. In view of economic considerations, it is increasingly necessary for the requirements to compromise between price and performance of the pigment to be used.
In the case of PETP attention must be paid to the nucleating effect of the organic pigments, depending on the application (bottles).
1.8.3.5 Polystyrene, Styrene-Copolymers, Poly(methyl methacrylate)
Polystyrene (PS), a highly rigid and surface-hardened thermoplastic, is glass clear and almost colourless. Its typical slight yellow tinge is easy to compensate for by adding transparent blue colourants to adjust the colour. Polystyrene softens between 80 and 100 °C. It is processed between about 170 and 280 °C, up to a maximum of 300 °C, without colour change, by any of the methods that are recommended for thermoplastics. The list of products includes extrusion made sheets, profiles and films, which are often foamed.
The mechanical properties of PS may be improved drastically by copolymerizing the monomer with one or more of various rubber-like materials (graft polymers). Impact resistant PS, for instance, contains 5–25% natural rubber, which is not dissolved but dispersed in PS. This and other dispersed additives, owing to a difference in refractive indices between plastic and filler, scatter much of the incident light and therefore afford opaque products; the degree of opacity in each individual case depends on the amount of rubber added. Impact resistant PS types are processed at 170–260 °C. Copolymers with acrylonitrile and butadiene have high impact strength and excellent fastness to ageing. The inherent colour of these ABS polymers is somewhat more yellowish than polystyrene; their high opacity is attributed to extensive light scattering. In recent years, transparent types of ABS have been marketed; these are processed at about 210–250 °C.
Poly(methyl methacrylate) (PMMA), an amorphous plastic material, is extremely stable to ageing and to weathering; it is hard and glass clear.
The transparency of a plastic, with polystyrene and styrene/acrylonitrile copolymer (SAN) being most transparent, to impact resistant PS, down to highly opaque ABS, clearly has some influence on the colouration of a product. The colouristic properties of a colourant depend on the plastic in which it is incorporated. In discussing the colouration of polymers whose glass transition temperature is far above room temperature, special rules have to be observed (Section 1.6.3). It is rarely possible for dissolved molecules to migrate, thus these polymers may be coloured with pigments and also soluble dyes. Some of these dyes are even acceptably light-fast and afford brilliant shades, especially in combination with opaque inorganic or organic pigments.
The requirements regarding heat stability are stringent for pigments that are used to colour these plastic materials. Few organic pigments tolerate the high end of the processing temperature range between 280 and 300 °C. Several types, however, withstand moderate temperatures between 220 and 260 °C.
Depending on the temperature, a large number of organic pigments dissolve more or less in this class of plastics. The colour almost invariably changes as the pigment dissolves, frequently accompanied by a change in the fastness properties, especially in the response of the system to light.
Completely dissolved pigments should be referred to as dyes and be tested as such. This concerns features such as the extraction properties in the finished article. In PS, SAN and other transparent plastics with a high glass transition temperature, they afford transparent, glass clear colourations.
Pigments that at low concentrations dissolve reasonably well at the temperature at which their medium is processed are frequently employed to advantage at higher concentrations. This phenomenon is readily explained by considering the influence of the dissolved versus the undissolved portion of pigment. The colouristic properties of the polymer will be enhanced if the undissolved pigment particles dominate over the dissolved portion in determining the colouristics of the system, often resulting in high brilliance. According to the laws of physical chemistry, organic pigments (and dyes) dissolve at a rate that is largely dependent not only on the pigment particle size but also on the available time. PS and its copolymers are normally processed as pellets, which may be coloured by colourants in powder, granulated or paste form. Pigment preparations for PS are also becoming increasingly important.
Pigments and pigment blends in powder form are incorporated in their medium by means of slow mixers. Organic pigments are often insufficiently wetted by molten PS or its copolymers and accordingly difficult to disperse. It is possible, however, to facilitate dispersion by initially applying an adhesion agent, that is, a wetting agent, in concentrations up to 0.3% relative to the granulated plastic. PS, being a brittle and hard material, may cause metal abrasion after a certain mixing time, which gives a dull effect to otherwise clean, brilliant shades; fluorescent colourants may even lose their fluorescence.
The advantage of using pigment preparations in paste form, which affords easy colour matching by blending the corresponding pastes, is compromised by the fact that a higher content of liquid component may affect the mechanical properties of the plastic and lead to stress corrosion cracking. Similarly, the applicability of pastes is restricted by physiological considerations. Used as carriers, for instance, they contain paraffin oil.
In exterior exposure, PS yellows somewhat, due to UV radiation. To shield the plastic from degradation in UV light, it is also supplied in combination with UV absorbents. This prolongs the lifetime of the products by a factor of three to five. Grades that contain UV absorbents are slightly yellowish, a fault that may be corrected by adding transparent blue colourants such as soluble dyes or Ultramarine Blue [270].
1.8.3.6 Elastomers
The characteristic property of elastomers is their rubber-elastic behaviour. Their softening temperature lies below room temperature. In the unvulcanized state, that is, without crosslinking of the molecular chains, elastomers are plastic and thermo-formable, but in the vulcanized state – within a certain temperature range – they deform elastically. Vulcanization converts natural rubber into the elastic state. Numerous synthetic rubber types and elastomers are known and available on the market. They have several specially improved properties over crude rubber – for instance substantially improved elasticity, heat, low-temperature, weathering and oxidation resistance, wear resistance, resistance to different chemicals or oils.
Pigments for colouring rubber compounds have to satisfy a range of requirements and, in particular, must not contain rubber poisons. Even small amounts of copper and manganese not only considerably impair vulcanization of the rubber but also cause accelerated ageing of the vulcanized rubber. Pigments for colouring rubber are therefore required to have a combined content of less than 0.01% of these two heavy metals. In the case of copper phthalocyanine blue pigments, somewhat higher values of non-complexed metal are accepted but under no circumstances more than 0.015%. Pigments intended for this use are often tested for their content of these metals.
The pigments are also required to have a specific heat resistance. This is tested on five colourations with different pigment concentrations in the range 0.01–1% together with ten-times the amount of chalk. The colourations placed side by side are vulcanized hot for 15 min at 140 °C and evaluated colouristically against the corresponding untreated comparison colouration.
In most cases the pigments used are also required to be migration resistant. The suitability test is also carried out with five different pigment concentrations. To test the fastness to bleeding, the unvulcanized colourations are brought into defined contact with a white milled sheet of specific composition and vulcanized wet for 20 min in open steam at 140 °C. During this process, half the colouration is often covered with a wet cotton cloth to determine whether the cloth, the rubber or both are stained by bleeding.
The resistance to blooming is tested by rubbing the colourations of differing concentrations with a white cloth immediately after they are produced, after storage for 6 months at room temperature and, if necessary, also in an accelerated test after storage for 24 h at 70 °C.
The weathering fastness of pigments for natural rubber is of virtually no significance because the material itself has poor weathering fastness. By contrast, some high-quality synthetic elastomers with high weathering fastness require a corresponding pigment. Resistance to specific chemicals is necessary in individual cases.
Rubber compounds are coloured with powder pigments and increasingly with granulated pigment preparations, known in the rubber industry too as master-batches.
1.8.3.7 Thermoplastic Elastomers (TPEs)
Thermoplastic elastomers are a group of material that in chemical structure and processing are between elastomers and thermoplasts. They posses rubber like properties but can be processed and recycled like regular polymers. There is a wide range of TPEs on the market, defined by their shore hardness (DIN EN ISO 868; DIN ISO 7619-1), ranging from Shore A10 to Shore D75.
Hereby, a hard phase is responsible for the strength and a soft phase for the softness. The phases are combined chemically or physically. The physical process is based on the blending/mixing of crosslinked or non-crosslinked elastomers mainly in polypropylene. The main types that are based on this process are TPE-O and TPE-V.
The chemical process is based on segmented blocks of different flexibility and hardness (Table 1.9). The main types for this process are TPE-S, TPE-U and TPE-E.
Table 1.9 Main types of thermoplastic elastomers.
| Group | Blends | Block copolymers |
| Structure | Two-phase system, mixture of crosslinked or non-crosslinked elastomers in mainly PP | Segmented blocks of different flexibility and hardness |
| Key types | TPE-O thermoplastic polyolefins with a non-crosslinked elastomer phase TPE-V thermoplastic vulcanized rubber = polyolefin with a crosslinked elastomer phase |
TPE-S styrene block copolymer TPE-U thermoplastic polyurethane elastomer TPE-E thermoplastic polyester elastomer TPE-A thermoplastic polyamide elastomer |

|

|
The largest group of the TPEs is TPE-S with a market share of around 50%, the second largest group is TPE-U (15%), followed by TPE-V (10%). In 2011 the global volume was around 3.48 mio t; the market growth is about 4.5% annually.
1.8.3.7.1 TPE-S: Styrene Block Copolymer – SBS, SEBS, SEPS
This is the largest group of the TPE family, based on at least three blocks, two styrene end blocks and one soft elastomeric block (polybutadiene, polyisoprene) in the middle. They are mainly processed by extrusion and injection moulding at processing temperatures of 180–250 °C.
Applications vary widely from ‘soft-touch’ children toys, automotive parts to food contact applications, for example in packaging or even wine bottle corks. This version is also used for replacing flexible PVC in medical applications.
1.8.3.7.2 TPE-U
The second largest group of the TPE family. TPE-U is a linear segmented block copolymer based on hard and soft segment. The hard segments can be either aromatic or aliphatic and are based on isocyanides. The soft segments can be polyether or polyester. This material is mainly processed by extrusion, injection-, blow- and compression-moulding at processing temperatures of 180–230 °C.
TPE-U can be found in sporting goods, shoe soles and as replacements for flexible PVC.
1.8.3.7.3 TPE-V/TPE-O
This group is based on a combination of polypropylene and ethylene–propylene (dien) rubber EP(D)M. TPE-V has a crosslinked elastomer phase and TPE-O, a non-crosslinked phase.
Owing to its advantage of excellent impact strength at low temperatures it is often used as an impact modifier; other applications can be found in the construction and automotive area.
1.8.3.7.4 Colouration of TPEs
Owing to the wide variability of the TPE family it is difficult to give a recommendation for the colouration. The pigment selection should be based on the heat stability and in particular bleeding fastness. In some TPEs, for example TPE-E (polyester type TPE) and TPE-U, some organic pigments like the quinacridones should be tested before commercial use due to their tendency to go into partial solution in those materials.
1.8.3.8 Thermosets (Thermosetting Plastics)
Thermosets are formed by crosslinking (curing) reactive linear and branched macromolecules and can be manufactured by polycondensation, polymerization and polyaddition. Thermosets can therefore be processed once only with the application of heat and pressure to form semi-finished products or finished articles and cannot be recovered; their processing is irreversible. Amongst the most familiar thermosets are the combinations of formaldehyde with phenol, resorcinol and so on (phenolics), urea, aniline, melamine and similar combinations (aminoplastics).
Because of the dark self-colour of phenolics, which are processed predominantly with a high filler content (up to 80%), there are limits to their colouration.
Thermosets are processed by compression moulding, transfer moulding and injection moulding or by extrusion, depending on their structure. The pressing temperatures are about 150–190 °C. Corresponding requirements are placed on the pigments. Dyes are often also used to colour thermosets.
The pigments are incorporated into the resins before crosslinking to ensure homogeneous colouration. This can be done in the molten resin, for example in a kneader at about 90 °C, or in dissolved or liquid resins by the liquid resin method. Ball mills are normally used for colouring pre-wetted powder moulding compounds that have not yet been cured.
The casting resins based on epoxy resins, methacrylate or unsaturated polyester are also thermosets. The epoxy resins are cured with amines or phthalic anhydride. Curing is not influenced by organic pigments. Moisture, on the other hand, delays curing. The pigments used must therefore be largely dry, which is generally the case.
Unsaturated polyester and methacrylate casting resins are normally cured with organic peroxides. The polyester casting resins are dissolved in monostyrene, and temperatures up to 200 °C are reached in their polymerization. Thick-walled articles in particular are often exposed to heat for a long period of time. The required heat resistance of the organic pigments used is governed by this.
Methacrylate resins are produced from monomeric methacrylic acid methyl ester, a markedly lower temperature being employed in their polymerization. The heat resistance of the pigments is therefore only of minor importance in this medium.
High light and weathering fastness is frequently required, for example in car body manufacture. These fastness properties can be severely impaired by the type and amount of the peroxide catalysts, and therefore peroxide-resistant pigments have to be used. At the same time they must not affect the curing process, that is, they must neither accelerate nor delay it. It is, however, known that organic pigments can behave completely differently, depending on the method of curing and type and amount of peroxide used [271]. Thus, under certain conditions dihydrazone yellow pigments such as Pigment Yellow 17 and 83 have no effect at all, whereas copper phthalocyanine green, for example, severely delays curing and copper phthalocyanine blue prevents it altogether. If another organic peroxide is used, however, curing is even accelerated slightly, for example in the case of copper phthalocyanine green.
Unsaturated polyester and methacrylate resins are frequently coloured with pigment–plasticizer (DIDP) pastes. They have no measurable adverse effect on the important mechanical properties of the finished article. To a small extent pigments are also dispersed directly in one part of the monomer.
1.8.3.9 Spin Dyeing
The technique of spin dyeing chemical fibres may be located somewhere between the textiles and the plastics area. In contrast to textile colouration, the material to be extruded is coloured before the fibre is made. The requirements to be met by pigments are therefore similar to those that apply to the colouration of plastics. Likewise, heat stability is the foremost concern in connection with pigment selection for spin dyeing. In addition, the pigment in this application must be insoluble in the solvents used.
The demands regarding pigment dispersibility are particularly high. Every care must be taken to ensure that the size of remaining pigment agglomerates does not exceed 2–3 µm [272, 273]. Larger particles adversely affect the tensile strength of the fibre and frequently cause failure through breakage, especially as the fibre is stretched. In practice, however, pigment powders rarely afford such a high degree of dispersion, and there is no guarantee for the quality of dispersion. It is therefore almost unavoidable to replace pigments by pigment preparations in spin dyeing processes. Clearly, a carrier material should be chosen that does not present any processing problems.
Three different methods are available for spin dyeing [274]:
- Melt spinning: This technique is used with thermoplastic material, such as polyester, polyamide or polypropylene. The polymer is melted in the extruder and then pressed through a spinnerette. It solidifies by cooling as it falls vertically through a shaft to the bottom of the extruder. Pigments are therefore expected to exhibit excellent heat stability. Regarding organic pigments, the spinning temperatures may vary over a wide range, depending on the melting point of the polymer (Table 1.10). As a rule, pigment preparations for this application are based on a carrier material that is identical or similar to the polymer to be extruded.
- Wet spinning: This technique is characterized by spinning a filtered viscous polymer mass, dissolved in a suitable solvent, into contact with a precipitation or coagulation bath. Polyacrylonitrile, poly(vinyl acetate), cellulose acetate and other materials are processed by this method. Thermal requirements for pigments are less stringent than for melt spinning, but pigments are expected to be fast to the solvents and chemicals used.
- Dry spinning: The polymer, dissolved in a suitable solvent and filtered, is pressed through spinnerettes and, in an oxygen-free atmosphere, pulled by vacuum through a heated shaft, where the polymer solidifies as the solvent evaporates. The requirements of this process regarding the heat stability of pigments are much less stringent than in melt spinning. However, like in wet spinning, pigments must be fast to the solvents used. Polyacrylonitrile, triacetate and polycarbonate are processed by this method.
Table 1.10 Melting points and spinning temperatures of various polymers.
| Polymer | Melting point (°C) | Spinning temperature (°C) |
| Polyester | 255 | 285 |
| PA 6 (6-polyamide) | 220 | 250–280 |
| PA 66 (nylon) | 245 | 275 |
| Polypropylene | 175 | 240–300 |
Colouration techniques have been introduced for several thermoplastics such as polyester [275–277]. Instead of using pigments, dyes are employed that dissolve in the polymer melt and are sufficiently heat stable and sublimation-fast to tolerate the processing conditions. These techniques have certain advantages over traditional methods based on organic pigments: they avoid fibre failure through breakage during stretching, plugging of the filter and other faults.
1.8.3.9.1 Polyacrylonitrile (PAC)
Polyacrylonitrile, which decomposes already at 220 °C, which is below its melting point (about 290 °C), cannot be processed by melt extrusion. Dry and wet spinning techniques are therefore used, and the list of appropriate solvents includes dimethylacetamide, dimethylformamide, dimethyl sulfoxide and aqueous solutions of inorganic salts. Besides being insoluble in the appropriate solvents, pigments that are to be used in speciality applications, such as window blinds, awnings and tents, should meet the respective demands regarding light fastness and weather fastness. Less lightfast pigments are acceptable for use in clothing, decorative and home textiles, that is, upholstery, curtains and carpeting. Polyacrylonitrile fibre is commonly used for these purposes and is quite adequate: it is by far the most weatherfast of all synthetic and natural fibres. Pigment preparations are also supplied for the colouration of PAC. Specialized cationic dyes are of negligible importance.
1.8.3.9.2 Polyester (PETP)
The thermal requirements for pigments targeted for PETP melt extrusion are particularly severe. However, it is important to consider the individual conditions at the various stages of polymer colouration. Pigments, for instance, that are added during the so-called condensation process in a glycol dispersion prior to transesterification or condensation in the autoclave are exposed to temperatures between 240 and 290 °C for 5–6 h [277]. These harsh conditions are tolerated by very few polycyclic pigments, primarily by representatives of the quinacridone, copper phthalocyanine, naphthalenetetracarboxylic acid, and perylene tetracarboxylic acid series.
The choice of pigments to be added to a ready-made polyester is much less restricted. At this stage, a pigment will only be exposed to heat for about 20–30 min, although the temperature will be equally high. This is made possible by mixing the granulated polyester with a pigment concentrate or with a pigment preparation, or by transferring the molten pigment concentrate to the melting zone of the spin extruder, for instance via a side-screw extruder. Clearly, the carrier material of the pigment preparation is equally subject to compatibility restrictions. In the case of polyester, the newer colouration technologies are carried out to advantage with dyes that are melt-soluble, sufficiently heat resistant, and sublimation proof during spinning.
1.8.3.9.3 Polyamide (PA)
Pigments targeted for polyamide spin dyeing, apart from being extremely heat stable, must also be chemically fast to the highly reducing medium of the PA melt. Spinning temperatures are between 250 and 290 °C, depending on the type of polyamide. As with polyester extrusion, very few polycyclic pigments are suitable for this purpose. There are no organic yellow pigments that satisfy the specifications. Pigment preparations are also available for this purpose.
1.8.3.9.4 Viscose
Viscose, the alkaline solution of sodium cellulose xanthate, is produced by treating cellulose with sodium hydroxide solution and carbon disulfide. Viscose is coloured in the form of cellulose xanthate prior to extrusion. Aqueous pigment preparations in paste form are employed. Apart from the usual fastness requirements, pigments are expected to be fast to strong acids, alkali and reducing agents. In addition, pigments that affect the so-called maturation of the viscose or the coagulation and regeneration in the spin bath or in one of the after-treatment baths are unacceptable. However, a considerable number of pigments satisfy these specifications [278].
1.8.4 Other Areas of Application
Organic pigments are also used to colour various materials outside the above-mentioned areas of printing inks (Section 1.8.1), paints (Section 1.8.2) and plastics and spin dyeing (Section 1.8.3). Pigment selection depends on the individual manufacturing and processing conditions, which define the demands placed on a pigment regarding solvent fastness and the performance of the ultimate product, especially its fastness to light.
1.8.4.1 Miscellaneous Applications
Areas of application include wood colouration [279], paper mass colouration [280] and paper surface coating in the lime press [281], the office articles and artists' colours sector; pigments are used in coloured pencils, crayons and writing and pastel chalks or in water colours, as well as in cosmetics, especially soap [282].
Printing on textiles is one of the major areas of pigment application and is usually considered separately from the graphics industry. Requirements to be met by pigments for this purpose depend especially on the expected performance of the final article, the printed textile (Section 1.6.2.4).
1.8.4.2 Colouration of Glass with Organic Pigments
Glass can generally not be coloured with organic pigments. The usual glass processing temperatures are so high that all organic compounds are decomposed. However, it was recently found that low-melting glass types (so-called solder glasses), with glass transition temperatures of 300–350°C can be coloured with organic pigments [283]. Application examples include phthalocyanine blue, perylene and perinone pigments. The pigment is mixed with the glass powder and heated to 300–350°C resulting in a mass-colouration of the glass by the organic pigment. The resulting glass is either directly used. Or the glass is powdered and subsequently used for the colouration of, e.g., polymeric materials.
Notes
References for Chapter 1
- 1 DIN 55943 (2001) Farbmittel, Begriffe. ISO (1984) ISO 4618-1-1984 (TC 35): Paints and varnishes–Vocabulary, Part 1: General terms.
- 2 Griesheim-Elektron (1911) DRP 251479
- 3 Geissler, G. (1977) DEFAZET Dtsch. Farben Z., 31, 152–156.
- 4 MacDonald Smith, H. (1977) Am. Ink Maker, 55, 6.
- 5 M. Klessinger, Chem. unserer Zeit 12 (1978) 1–11; further references ibid.; Dähne, S. (1970) Z. Chem., 10, 133, 168; Dähne, S. (1978) Science, 199 (4334), 1163–1167.
- 6 Rys, P. and Zollinger, H. (1982) Farbstoffchemie, vol. 3. Suppl., Verlag Chemie, Weinheim; Zollinger, H. (2006) Color Chemistry, 3rd edn, VCH Verlagsgesellschaft, Weinheim.
- 7 Fabian, J. and Hartmann, H. (1980) Light Absorption of Organic Colorants, Springer-Verlag, Berlin.
- 8 Griffiths, J. (1981) Rev. Prog. Color., 11, 37–57.
- 9 Griffiths, J. (1976) Color and Constitution of Organic Molecules, Academic Press, London.
- 10 Lincke, G. (1980) Farbe+Lack, 86, 966–972.
- 11 Eulitz, G. and Hoechst A.G., personal communication.
- 12 Christie, R.M. and Standring, P.N. (1989) Dyes Pigments, 11, 109–121.
- 13 Lincke, G. (1970) Farbe+Lack, 76, 764–774.
- 14 Gläser, F. (1976) XIII. Congr. FATIPEC, Juan les Pins, 1976, Congress book, 239–243.
- 15 Paulus, E.F. and Hunger, K. (1980) Farbe+Lack, 86, 116–120.
- 16 Hunger, K., Paulus, E.F., and Weber, D. (1982) Farbe+Lack, 88, 453–458.
- 17 For example, Allen, T. (1968) Particle Size Measurement, Chapman & Hall, London.
- 18 DIN 53 206 (1972) Teil 1: Teilchengrößenanalyse, Grundbegriffe.
- 19 Brunauer, S., Emmet, P.H., and Teller, E. (1938) J. Am. Chem. Soc., 60, 309–319; Brunauer, S. (1943) The Adsorption of Gases and Vapours, Oxford University Press, London.
- 20 de Boer, J.H. (1958) Structure and Properties of Porous Materials (eds D.H. Everett and F.S. Stone), Butterworth, London.
- 21 Everett, D.H. and Ottewill, R.H. (eds.) (1970) Surface Area Determination, Butterworth, London.
- 22 Wedler, C. (1970) Adsorption, Verlag Chemie, Weinheim.
- 23 DIN 66 131 (1993) Bestimmung der spezifischen Oberfläche von Feststoffen durch Gasadsorption nach Brunauer, Emmet and Teller (BET), see also ISO 9277–1992.
- 24 DIN 66 132 (1975) Bestimmung der spezifischen Oberfläche von Feststoffen durch Stickstoffadsorption.
- 25 Robens, E. (1969) Lab. Pract., 18, 292; Gregg, S.J. and Sing, K.S.W. (1967) Academic Press, New York, p. 330.
- 26 Giles, C.H. and Nakhwa, S.N. (1962) J. Appl. Chem., 12, 266.
- 27 Herrmann, M. and Honigmann, B. (1969) Farbe+Lack, 75, 337–342.
- 28 Merkle, K. and Herbst, W. (1976) Farbe+Lack, 82, 7–14.
- 29 List, K.H. and Weissert, J. (1964) Chem. Ing. Techn., 36, 1051–1053.
- 30 Carman, P.C. (1938) J. Soc. Chem. Ind. (London), 57, 225 and 58 (1939) 1; see also DIN 66 126, p. 1.
- 31 DIN 66 127, Bestimmung der spezifischen Oberfläche pulverförmiger Stoffe mit Durchströmungsverfahren; Verfahren and Gerät nach Blaine.
- 32 DIN 53 199, Bestimmung der Ölzahl (Spatelverfahren). ISO 787 T5: General methods of testing for pigments and extenders, Part 5: Determination of oil absorption value.
- 33 Beddow, J.K. (1980) Particulate Science and Technology, Chemical Publishing Co. Inc., New York.
- 34 Irani, R.R. and Callis, F.C. (1963) Particle Size: Measurement, Interpretation and Application, John Wiley & Sons, Inc., New York.
- 35 Cadle, R.D. (1965) Particle Size, Reinhold, New York.
- 36 Maikowski, M.A. (1976) Prog. Colloid Polym. Sci., 59, 70–81.
- 37 Beresford, J. (1967) J. Oil Color Chem. Assoc., 50, 594–614. Joyce-Loebl Ltd. Technical Operations Inc. Burlington Mass., USA.
- 38 Hauser, P. and Honigmann, B. (1977) Farbe+Lack, 83, 886–890.
- 39 Graphisches Tablett, MOP, halbautomatisches Bildanalyse-System. Manufactured by Fa. Kontron, Augsburg
- 40 Völz, H. and Weber, G. (1984) XVII. Congr. FATIPEC, Lugano 1984, Congress book, Vol. IV, 77–90; Farbe + Lack, 90, 642–646.
- 41 DIN 55 302 (1967) Häufigkeitsverteilung, Mittelwert and Streuung.
- 42 DIN 66 141 (1974) Darstellung von Korn-(Teilchen-)größenverteilungen: Grundlagen. ISO 9276-1-1988: Representation of particle size analysis, Part 1: Graphical representation.
- 43 DIN 66 143 (1974) Potenznetz.
- 44 DIN 66 144 (1974) Logarithmisches Normalverteilungsnetz.
- 45 DIN 66 145 (1976) RRSB-Netz.
- 46 Paulus, E.F. (1980) Ullmanns Enzyclopädie der technischen Chemie, 4th edn, vol. 5, VCH Verlagsgesellschaft, Weinheim, pp. 235–268.
- 47 Von Susich (1948), FIAT Final Report No. 1313, “German dyestuffs and dyestuff intermediates, including manufacturing processes, plant design, and research data”, Vol. 3: “Dyestuff research”, prepared by Field Information Agency, Technical (FIAT), United States group control council for Germany, p. 412-463; which is a translation of an unpublished report on the application of the X-ray technique in the chemical industry by Dr. Georg von Susich of Wilhelmsfeld near Heidelberg relating to the work of von Susich at the I.G. laboratories in Ludwigshafen. Page 437; in addition, p. 441.
- 48 Ogawa, T., Kuwamoto, K., Isoda, S., Kobayashi, T., and Karl, N. (1999) Acta Cryst. B, 55, 123–130.
- 49 Fukushima, K., Nakatsu, K., Takahashi, R., Yamamoto, H., Gohda, K., and Homma, S. (1998) J. Phys. Chem. B, 102, 5985–5990.
- 50 Hunger, K. (1999) Rev. Prog. Coloration, 29, 71–84.
- 51 Paulus, E.F., Leusen, F.J.J., and Schmidt, M.U. (2007) CrystEngComm, 9, 131–143.
- 52 Schmidt, M.U., Brühne, S., Wolf, A.K., Rech, A., Brüning, J., Alig, E., Fink, L., Buchsbaum, C., Glinnemann, J., van de Streek, J., Gozzo, F., Brunelli, M., Stowasser, F., Gorelik, T., Mugnaioli, E., and Kolb, U. (2009) Acta Crystallogr. B, 65, 189–199.
- 53 Klebe, G., Graser, F., Hädike, E., and Berndt, J. (1989) Acta Cryst. B, 45, 69–77.
- 54 Mizuguchi, J., Hino, K., and Tojo, K. (2006) Dyes Pigments, 70, 126–135.
- 55 Schmidt, M.U., Dinnebier, R.E., and Kalkhof, H. (2007) J. Phys. Chem. B, 111, 9722–9732.
- 56 Paulus, E.F. and Rieper, W. (1985) Z. Kristallogr., 171, 87–100.
- 57 van de Streek, J., (2015) Acta Crystallogr., Sect. B., 71, in press.
- 58 Ivashevskaya, S.N., van de Streek, J., Djanhan, J.E., Brüning, J., Alig, E., Bolte, M., Schmidt, M.U., Blaschka, P., Höffken, H.W., and Erk, P. (2009) Acta Crystallogr., Sect. B, 65, 212–222.
- 59 Schmidt, M.U., van de Streek, J., and Ivashevskaya, S.N. (2009) Chem. Eur. J., 15, 338–341.
- 60 McCrone, W.C. (1965) Physics and Chemistry of the Organic Solid State, vol. 2 (eds D. Fox, M.M. Labes, and A. Weissberger), Interscience Publishers, London, pp. 725–767.
- 61 Schmidt, M.U. and Metz, H.J. (Clariant) (1999) EP 965616.
- 62 Schmidt, M.U. and Metz, H.J. (Clariant) (1999) EP 965617.
- 63 Schmidt, M.U. (Clariant) (1999) EP 1010732.
- 64 Erk, P., Hengelsberg, H., Haddow, M.F., and van Gelder, R. (2004) CrystEngComm, 6, 474–483.
- 65 Erk, P. (BASF), personal communication.
- 66 Jung, R., Klaus, K., Nestler, B., Schmidt, M.U., Unverdorben, L. (Clariant), and Steiner, R. (2000) EP 1240253.
- 67 Paulus, E.F. (1992) Ullman's Encyclopedia of Industrial Chemistry, 5th edn, vol. 20, VCH Verlagsgesellschaft, Weinheim, p. 412ff.
- 68 Mizuguchi, J. (1981) Cryst. Res. Techn., 16, 695–700.
- 69 van de Streek, J., Brüning, J., Ivashevskaya, S.N., Ermrich, M., Paulus, E.F., Bolte, M., and Schmidt, M.U. (2009) Acta Crystallogr., Sect. B, 65, 200–211.
- 70 Schmidt, M.U., Ermrich, M., and Dinnebier, R.E. (2005) Acta Crystallogr., Sect. B, 661, 37–45.
- 71 Radtke, W., Erk, P., and Sens, B. (2009) High Performance Pigments, 2nd edn, (eds E. Faulkner and R. Schwartz), Wiley-VCH, Weinheim, pp. 221–241.
- 72 Schmidt, M.U., Hofmann, D.W.M., Buchsbaum, C., and Metz, H.J. (2006) Angew. Chem., 118, 1335–1340; Angew. Chem. Int. Ed., 45, 1313–1317.
- 73 Schmidt, M.U. and Dinnebier, R.E. (1999) J. Appl. Cryst., 32, 178–186.
- 74 Bekoe, S.L., Hammer, S.M., and Schmidt, M.U. (2012) Angew. Chem., 124, 4814–4818.
- 75 Gorelik, T., Schmidt, M.U., Brüning, J., Bekö, S., and Kolb, U. (2009) Crystal Growth & Design, 9, 3898–3903.
- 76 Neumann, M.A., Leusen, F.J.J., and Kendrick, J. (2008) Angew. Chem., 120, 2461–2464.
- 77 Schmidt, M.U. and Englert, U. (1996) J. Chem. Soc., Dalton Trans., 1996, 2077–2081.
- 78 Schmidt, M.U. (1999) in Crystal Engineering: From Molecules and Crystals to Materials, NATO Science Series, Series C, vol. 538 (eds D. Braga, F. Grepioni and A.G. Orpen), Kluwer Academic Publishers, Dordrecht, pp. 331–348.
- 79 Schmidt, M.U., Wacker, A., and Metz, H.J. (Clariant) (2004), EP 1659336.
- 80 Judd, D.B. and Wyczecki, G. (1975) Color in Business, Science and Industry, John Wiley & Sons, Inc., New York; Richter, M. (1976) Einführung in die Farbmetrik, Walter de Gruyter, Berlin; Hunter, R.S. (1975) The Measurement of Appearance, John Wiley & Sons, Inc., New York.
- 81 Völz, H.G. (1990) Industrielle Farbprüfung. Grundlagen und Methoden, VCH Verlagsgesellschaft, Weinheim; Völz, H.G. (1994) Industrial Color Testing. Fundamentals and Techniques, VCH Verlagsgesellschaft, Weinheim; Lang, H. (1993) Bergmann-Schäfer, Lehrbuch der Experimentalphysik, vol. 3, Optik, 9. ed., Walter de Gruyter, pp. 665–745.
- 82 DIN 5033-3 1979 (1980) Teil 1, Grundbegriffe der Farbmetrik. ISO 7724-1-1984: CIE Standards colorimetric observers; Paints and varnishes – Colorimetry, Part 1: Principles: ISO 7724-2-1984, Part 2: Color measurement.
- 83 DIN 6174 (1979) Farbmetrische Bestimmung von Farbabständen bei Körperfarben nach der CIELAB-Formel. ISO 7724-3-1984: Paints and varnishes – Colorimetry, Part 3: Calculation of color differences, 1. ed.
- 84 Raabe, P. and Koch, O. (1957) Melliand Textilber., 38, 173.
- 85 Geißler, G., Schmelzer, H., and Müller-Kaul, W. (1978) Farbe+Lack, 84, 139.
- 86 DIN 53 235-3-1977 (1977) Prüfungen an standardfarbtiefen Proben.
- 87 Duncan, D.R. (1940) Proc. Phys. Soc. London, 52, 380.
- 88 Hoffmann, K. (1970) Farbe+Lack, 76, 665.
- 89 DIN 6164-3 (1981) DIN-Farbenkarte.
- 90 DIN 54 001-8-1982 (1982) Herstellung und Handhabung des Graumaßstabes zur Bewertung der Änderung der Farbe. ISO 105-A02-1993. prEN 20 105-A02-1991.
- 91 DIN 55 987-2-1981 (1981) Bestimmung eines Deckvermögenswertes pigmentierter Medien; Farbmetrisches Verfahren. ISO 6504-5-1987: (TC 35): Determination of hiding power. Part 5: Color difference method for dark colored paints.
- 92 DIN 16 524 T1, Widerstandsfähigkeit gegen verschiedene physikalische und cemische Einflüsse.
- 93 DIN 53 197, Bestimmung des Gehaltes an wasserlöslichen Anteilen. ISO 787-8-1983: General methods of test for pigments and extenders, Part 8: Determination of matter soluble in water – cold extraction method.
- 94 DIN 53202, Bestimmung der Aciditätszahl oder Alkalitätszahl. ISO 787-4-1981: General methods of test for pigments and extenders, Part 4: Determination of acidity or alkalinity of the aqueous extract.
- 95 DIN 53 200, Bestimmung des pH-Wertes von wäßrigen Pigment-Suspensionen. ISO 787-9-1985: General methods of test for pigments and extenders, Part 9: Determination of pH value of aqueous pigment suspensions.
- 96 DIN 53 208, Bestimmung der elektrischen Leitfähigkeit und des spezifischen Widerstandes von wäßrigen Pigment-Extrakten. ISO 787-14-1985: General methods of test for pigments, Part 14: Determination of resistivity of aqueous extract.
- 97 DIN 53 207 TI, T2, Bestimmung des Gehaltes an wasserlöslichen Sulfaten, Chloriden und Nitraten. ISO 787-13-1984: General methods of testing for pigments, Part 13: Determination of water-soluble sulfates, chlorides and nitrates.
- 98 DIN 16 524: Prüfung von Drucken und Druckfarben des graphischen Gewerbes.
- 99 Empfehlung des Bundesgesundheitsamtes (BGA) IX B II vom 1.7.72. (Recommendation of the German Federal Health Office).
- 100 DIN 54 002 (1982) Herstellung und Handhabung des Graumaßstabes zur Bewertung des Anblutens. ISO 105-A03-1993: Textiles – Tests for color fastness, Part A03: Grey scale for assessing staining.
- 101 DIN 54 005, DIN 54 006 (1983) Bestimmung der Wasserechtheit von Färbungen und Drucken (leichte bzw. schwere Beanspruchung). ISO 105-E01-1989: Textiles – Tests for color fastness, Part E01: Color fastness to water. prEN 20 105-E01-1992.
- 102 DIN 54 007 (1983) Bestimmung der Meerwasserechtheit von Färbungen und Drucken. ISO 105-E02-1989: Textiles for color fastness, Part E02: Color fastness to sea water. prEN 20 105-E02-1992.
- 103 DIN 54 010–DIN 54 014 (1983) Bestimmung der Waschechtheit von Färbungen und Drucken. ISO 105-C03-1989: Textiles for color fastness, Part C03: Color fastness to washing: Test 3. ISO 105-C04-1989, Part C04: Color fastness to washing: Test 4; DIN EN 20 105-C04-1993. ISO 105-C05-1989, Part C05: Color fastness to washing: Test 5; DIN EN 20 105-C05-1993. ISO 105-C02-1989, Part C02: Color fastness to washing: Test 2; DIN EN 20 105-C02-1993. ISO 105-c1-1989, Part c1: Color fastness to washing: Test 1. DIN EN 20 105-c1-1993.
- 104 DIN 54 015, Bestimmung der Peroxid-Waschechtheit von Färbungen und Drucken.
- 105 DIN 54 016, Bestimmung der Hypochlorit-Waschechtheit von Färbungen und Drucken.
- 106 DIN 54 019 (1984) Bestimmung der Farbechtheit von Färbungen und Drucken gegenüber gechlortem Wasser. ISO 105-E03-1987: Textiles for color fastness, Part E03: Color fastness to chlorinated water (swimming-bath water).
- 107 DIN 54 020 (1983) Bestimmung der Schweißechtheit von Färbungen und Drucken. ISO 105-E04-1989: Textiles for color fastness, Part E04: Color fastness to perspiration. prEN 20 105-E04-1992.
- 108 DIN 54 021 (1984) Bestimmung der Reibechtheiten von Färbungen und Drucken. ISO 105X12-1987: Textiles – Tests for color fastness, Part X12: Color fastness to rubbing. prEN 20 105-X12-1992.
- 109 DIN 54 022 (1984) Bestimmung der Bügelechtheit von Färbungen und Drucken. ISO 105-X11-1987: Textiles – Tests for color fastness, Part X11: Color fastness to hot pressing. prEN 20 105-X11-1992.
- 110 DIN 54 024 (1984) Bestimmung der Trockenreinigungsechtheit von Färbungen und Drucken. ISO 105-D01-1987: Textiles – Tests for color fastness, Part D01: Color fastness to dry cleaning. prEN 20 105-D01-1992.
- 111 DIN 54 023 (1984) Bestimmung der Lösungsmittelechtheit von Färbungen und Drucken. ISO 105-X05-1987: Textiles – Tests for color fastness, Part X05: Color fastness to organic solvents.
- 112 DIN 54 028 (1984) Bestimmung der Säureechtheit von Färbungen und Drucken. ISO 105-E05-1989: Textiles – Tests for color fastness, Part E05: Color fastness to spotting: Acid.
- 113 DIN 54 030 (1984) Bestimmung der Alkaliechtheit von Färbungen und Drucken. ISO 105-E06-1989: Textiles – Tests for color fastness, Part E06: Color fastness to spotting: Alkali.
- 114 DIN 54 031 (1984) Bestimmung der Sodakochechtheit von Färbungen und Drucken. ISO 105-X06-1989: Textiles – Tests for color fastness, Part X06: Color fastness to soda boiling.
- 115 DIN 54 033–DIN54 037 (1984) Bestimmung der Peroxid-, Hypochlorit-, Chlorit-Bleichechtheit (leichte bzw. schwere Beanspruchung). ISO 105-N02-1993: Textiles, Tests for color fastness, Part N02: Color fastness to bleaching agencies.
- 116 DIN 54 060 (1985) Bestimmung der Trockenhitzeplissier- und Trockenhitzefixierechtheit von Färbungen und Drucken. ISO 105-P01-1993: Textiles – Tests for color fastness, Part P01: Color fastness to heat treatments. prEN 20 105-P01-1992.
- 117 Gläser, F. (1974) XII. Congres FATIPEC, Garmisch, 1974, Congress book, 363–370.
- 118 McDowell, W. (1972) J. Soc. Dyers Color., 88, 212–216.
- 119 DIN 53 775, Prüfung von Pigmenten in PVC weich.
- 120 Kumis, C.A. and Roteman, J. (1961) J. Polym. Sci., 55, 699.
- 121 Herbst, W. and Mettle, K. (1969) Farbe+Lack, 75, 1137–1157.
- 122 DIN 55 943 (1984) Farbmittel; Begriffe. ISO 4618-1-1984 and ISO 4618-2-1984: Paints and varnishes – Vocabulary, Part 1: General terms; Part 2: Terminology relating to initial defects and to undesirable changes in films during ageing.
- 123 DIN 55 945 (1988) Anstrichstoffe und ähnliche Beschichtungsstoffe; Begriffe. ISO 4618-4-1988: Paints and varnishes–Vocabulary, Part 4: Further general terms and terminology relating to changes in films and raw materials.
- 124 DIN 53 405 (1981) Bestimmung der Wanderung von Weichmachern. ISO 177-1988: Plastics – Determination of migration of plasticizers.
- 125 DIN 53 991 (1985–1988) Bestimmung des Ausblutens.
- 126 DIN 53 415 (1989) Bestimmung des Ausblutens von Farbmitteln. ISO 183-1976: Plastics – Qualitative evaluation of the bleeding of colorants.
- 127 Richter, J. (1968) Plastverarbeiter, 18, 933–935.
- 128 Memmel, F. (1971) Plastverarbeiter, 21, 325–328.
- 129 Völz, H.G., Kämpf, G., and Fitzky, H.G. (1970) X. Congr. FATIPEC, Montreux, 1970, Congress book, 107–112.
- 130 Kämpf, G., Papenroth, W., and Holin, R. (1972) XI. Congr. FATIPEC, Florenz, 1972, Congress book, 569–574.
- 131 DIN 53 159 (1999) Bestimmung des Kreidungsgrades von Anstrichen und ähnlichen Beschichtungen nach Kämpf.
- 132 DIN 53 223, Bestimmung des Kreidungsgrades nach der Klebebandmethode. ISO 4628-6-1990: Paints and varnishes – Evaluation of degradation of paint coatings – Designation of intensity, quantity and size of common types of defect, Part 6: Designation of degree of chalking.
- 133 Johnsen, U. and Moos, K.-H. (1978) Angew. Makromol. Chem. 74, 1–15.
- 134 Woebcken, W. and Seus, E. (1967) Kunststoffe, 57, 637–644, 719–723.
- 135 Rumpf, H. (1965) Chem.-Ing. Techn., 37, 187.
- 136 Pahlke, H. (1966) Farbe Lack, 72, 623–630, 747–758.
- 137 Gall, L. and Kaluza, U. (1975) DEFAZET Dtsch. Farben Z., 29, 102–115.
- 138 Neumann, A.W. and Sell, P.J. (1969) Z. Phys. Chem., 187, 227.
- 139 Hellwig, G. and Neumann, A.W. (1967) Farbe+Lack, 73, 823.
- 140 Schröder, J. and Honigmann, B. (1981) Farbe+Lack, 87, 176–180; Schröder, J. (1985) Farbe+Lack, 91, 11–17.
- 141 Sell, P.J. and Renzow, D. (1975) Progr. Org. Coat., 3, 323–348; (1977) Farbe+Lack, 83, 265–269.
- 142 Wolf, K.L. (1957) Physik und Chemie der Grenzflächen, Bd. I, Springer-Verlag, Berlin.
- 143 Patton, T.C. (1964) Paint Flow and Pigment Dispersion, John Wiley & Sons, Inc., New York.
- 144 Washburn, E.W. (1921) Phys. Rev., 17, 273.
- 145 Parfitt, G.D. (1967) J. Oil Color Chem. Assoc., 50, 826.
- 146 Herbst, W. (1972) DEFAZET Dtsch. Farben Z., 26, 519–532, 571–576.
- 147 Herbst, W. (1970) Farbe+Lack, 76, 1190–1208.
- 148 Beeferman, H.L. and Bergren, D.A. (1966) Paint Technology, 38, 492–500.
- 149 Herbst, W. (1972/73) Progr. Org. Coat., 1, 267–331.
- 150 Quednau, P. (1982) Polym. Paint Color, 18, 515–520.
- 151 Kaluza, U. (1979) DEFAZET Dtsch. Farben Z., 33, 355–359, 399–411.
- 152 Deryaguin, B.V. and Landau, L.D. (1941) Acta Physicochim. URSS, 14, 633.
- 153 Verwey, E.J.W. and Overbeck, J.Th.G. (1948) Theory of the Stability of Lyophobic Colloids, Elsevier, Amsterdam.
- 154 Napper, D.H. (1977) J. Colloid Interface Sci., 58, 390.
- 155 Crowl, V.T. (1967) J. Oil Color Chem. Assoc., 50, 1023.
- 156 Clayfield, E.J. and Lumb, E.C. (1966) J. Colloid Interface Sci., 22, 269.
- 157 Topham, A. (1977) Progr. Org. Coat., 5, 237.
- 158 Asbeck, W.K. and van Loo, M. (1949) Ind. Eng. Chem., 41, 1470.
- 159 Asbeck, W.K., Scherer, G.A., and van Loo, M. (1955) Ind. Eng. Chem., 47, 1472–1476.
- 160 Literature references see Kolár, O., Svoboda, B., Hájek, F. and Korinsky, J. (1969) Farbe+Lack, 75, 1039; Fürbeth, W., Grundmeier, G. and Stratmann, M. (1996) Farbe+Lack, 102, 78.
- 161 Merkle, K. and Herbst, W. (1971) Farbe+Lack, 77, 214–223.
- 162 Herbst, W. and Merkle, K. (1978) Farbe+Lack, 78, 25–34.
- 163 DIN ISO 1524-6 (1987) and EN 21 524, Lacke und Anstrichstoffe – Bestimmung der Mahlfeinheit (Körnigkeit). Determination of fineness of grind. Determination de la finesse de broyage.
- 164 DIN ISO 8781-2 (1992) Pigmente und Füllstoffe; Verfahren zur Beurteilung des Dispergierverhaltens. Bestimmung der Mahlfeinheit (Körnigkeit), Part 2: Assessment from the change in the fineness of grind.
- 165 DIN ISO 8780, Dispergierverfahren zur Beurteilung des Dispergierverhaltens; Methods of dispersion of assessment of dispersion characteristics; DIN ISO 8780-1 (1992) Dispersion characteristics of pigments and extenders – Methods of dispersion; DIN ISO 8780-2 (1990) Dispergieren mit einer Schüttelmaschine. Dispersion using an oscillatory shaking machine; DIN ISO 8780-3 (1990) Dispergieren mit einem Hochgeschwindigkeitsrührer; DIN ISO 8780-4 (1990) Dispergieren mit einer Perlmühle; DIN ISO 8780-5 (1990) Dispergieren mit einer Teller-Farbenausreibmaschine. Dispersion using an automatic miller; DIN ISO 8780-6 (1990) Dispergieren mit einem Dreiwalzwerk. Dispersion using a triple roll mill.
- 166 DIN 53 238 T 30 (1978) Prüfmedium Alkydharzsystem, niedrig-viskos, oxidativ trocknend; DIN 53238 T 31 (1978) Prüfmedium Alkyd/Melaminharz-System, niedrig-viskos, ofentrocknend; DIN 53238 T 32 (1982) Prüfmedium Alkyd/Melaminharz-System 2, niedrig-viskos, ofentrocknend; DIN 53238 T 33 (1984) Prüfmedium hochviskos, oxidativ trocknend.
- 167 Klaeren, A. and Völz, H.G. (1975) Farbe+Lack, 81, 709.
- 168 Pigenot, D.v. (1964) VII. Congr. FATIPEC 1964, Congress book, 249.
- 169 Zorll, U. (1974) Farbe+Lack, 80, 17.
- 170 Schmitz, O.J., Kroker, R., and Pluhar, P. (1973) Farbe+Lack, 79, 733.
- 171 DIN ISO 8781-1 1992 (1992) Verfahren zur Beurteilung des Dispergierverhaltens; Bestimmung der Farbstärkeentwicklung von Buntpigmenten. Assessment from the change in tinting strength of colored pigments.
- 172 DIN ISO 8781-3 (1992) Verfahren zur Beurteilung des Dispergierverhaltens; Bestimmung der Glanzentwicklung. Assessment from the change in gloss.
- 173 DIN 67 530-1982 (1982) Reflektometer als Hilfsmittel zur Glanzbeurteilung an ebenen Aufstrich- und Kunststoff-Oberflächen. ISO 2813-1989.
- 174 Gomm, A.S., Hull, G., and Moilliet, J.L. (1968) J. Oil Color Chem. Assoc., 51, 143–160.
- 175 White, J.W. (1976) Am. Paint Coat. J., 61, 52–56.
- 176 DIN 54 003 (1983) Bestimmung der Lichtechtheit von Drucken und Färbungen mit Tageslicht. ISO 105-B01-1993: Textiles; Tests for color fastness; Color fastness to light: Daylight.
- 177 DIN 16 525 (1965) Prüfen von Drucken und Druckfarben des graphischen Gewerbes; Lichtechtheit.
- 178 Schmelzer, H. (1984) XVII. Congr. FATIPEC, Lugano, 1984, Congress book, Vol. 1, 499–509.
- 179 Boxhammer, J. (1977), VDI-Seminar «Alterung von Kunststoffen», Original Hanau Quarzlampen GmbH, Hanau.
- 180 DIN 53 387 (1989) Kurzprüfung der Lichtechtheit. ISO 4892-1989: Plastics – Methods to exposure to laboratory light sources.
- 181 DIN 1249, Fensterglas.
- 182 DIN 53 236 (1983) Meß- und Auswertbedingungen zur Bestimmung von Farbunterschieden bei Anstrichen, ähnlichen Beschichtungen und Kunststoffen. ISO 7724-2-1984: Paints and varnishes – Colorimetry, Part 2: Color measurement.
- 183 DIN 53 387 (1989) Kurzprüfung der Wetterbeständigkeit (Simulation der Freibewitterung durch gefilterte Xenonbogen- Strahlung und Beregnung). ISO 4892-1-1994, ISO 4892-2-1994: Methods to exposure to laboratory light sources.
- 184 Kämpf, G., Papenroth, W., and Holm, R. (1973) Farbe+Lack, 79, 9–21.
- 185 Baier, E. (1977) Farbe+Lack, 83, 599–610.
- 186 Herbst, W. and Hafner, O. (1976) Farbe+Lack, 82, 394–411.
- 187 DIN 53 772 (1988) Bestimmung der Hitzebeständigkeit durch Spritzgießen.
- 188 DIN 24 450 (1987) Maschinen zum Verarbeiten von Kunststoffen.
- 189 DIN 53 774 (1986) Prüfung von Farbmitteln in PVC hart.
- 190 DIN 53 775, Prüfung von Pigmenten in PVC weich.
- 191 Hauser, P., Hermann, M., and Honigmann, B. (1970) Farbe+Lack, 76, 545–550; Hauser, P., Hermann, M. and Honigmann, B. (1971) Farbe+Lack, 77, 1097–1106.
- 192 Fuchs, O. (1968) DEFAZET Dtsch. Farben Z., 22, 548; Fuchs, O. (1969) DEFAZET Dtsch. Farben Z., 23, 17, 57, 111.
- 193 Zorll, U. (1980) Farbe+Lack, 86, 301–307.
- 194 Kaluza, U., (1979) Physikalisch-chemische Grundlagen der Pigmentverarbeitung für Lacke und Druckfarben, BASF Aktiengesellschaft.
- 195 Pahlke, H. (1970) X. Congr. FATIPEC, Montreux, 1970, Congress book, 549–554.
- 196 South, G.R. (1968) Am. Ink Maker, 32, 35–37.
- 197 DIN 16 5151/1: Farbbegriffe im Graphischen Gewerbe.
- 198 Zettlemoyer, A.C., Scarr, R.F., and Schaeffer, W.D. (1957) Proc. TAGA, 9A, 75; Zettlemoyer, A.C., Scarr, R.F. and Schaeffer, W.D. (1958) Int. Bull. Print Allied Trades, 80, 88–96.
- 199 Herbst, W. (1969) Farbe+Lack, 75, 431–436.
- 200 Schurz, J. and Kashmoula, T. (1976) Farbe+Lack, 82, 895–901.
- 201 See, e.g., publications of Anton Paar KG, Graz.
- 202 DIN 53 019 T1 (1980) Messung von Viskositäten und Fließkurven mit Rotationsviskosimetern mit Standardgeometrie. ISO 8961-1987: Plastics – Polymer dispersions; Definition and determination of properties.
- 203 Patton, T.C. (1966) J. Paint Technol., 38, 656–666.
- 204 Snell, F.D. and Hilton, C.L. (eds) (1966) Encyclopedia of Industrial Chemical Analysis, vol. 3, John Wiley & Sons, Inc., pp. 408–463.
- 205 DIN 53 018/1 (1976) Messung der dynamischen Viskosität newtonscher Flüssigkeiten mit Rotationsviskosimetern; Grundlagen.
- 206 DIN 53 018/2 (1976) Fehlerquellen and Korrekturen bei Zylinder-Rotationsviskosimetern.
- 207 DIN EN ISO 2884-1 (2006–2009) Paints and varnishes – Determination of viscosity using rotary viscometers.
- 208 DIN 53 222 (1990) Bestimmung der Viskosität mit dem Fallstabviskosimeter.
- 209 Schurz, J. (1972) Viskositätsmessungen an Hochpolymeren, Verlag Berliner Union Kohlhammer, Stuttgart.
- 210 Plajer, O. (1978) Plastverarbeiter, 29, 169–175, 249–252, 311–314, 376–378.
- 211 Schmelzer, H. (1973) Farbe+Lack, 79, 1066–1071.
- 212 Gall, L. (1970) Farbmetrik auf dem Pigmentgebiet, BASF AG, Ludwigshafen.
- 213 Maikowski, M.A. (1970) Melliand Textilber., 574–578.
- 214 Maikowski, M.A. (1967) Ber. Bunsenges. Phys. Chem., 71, 313–326.
- 215 DIN 6164, DIN-Farbenkarte.
- 216 Hauser, P., Horn, D., and Sappok, R. (1974) XIII. Congr. FATIPEC, Juan les Pins, 1974, Congress book, 191.
- 217 Sappok, R. (1978) J. Oil Color Assoc., 61, 299.
- 218 Gläser, F. (1978) DEFAZET Dtsch. Farben Z., 32, 338–342.
- 219 Zorll, U. (1966) Farbe+Lack, 72, 733–742.
- 220 Herbst, W. (1973) J. Paint Technol., 45, 39–50.
- 221 Gall, L. (1966) Farbe+Lack, 72, 955–965.
- 222 DIN 55 987 (1981) Bestimmung eines Deckvermögenswertes pigmentierter Medien; Farbmetrisches Verfahren. ISO 6504-5-1987 (TC35): Determination of hiding power, Part 5: Color difference method for dark colored paints.
- 223 Völz, H.G. (1976) DEFAZET Dtsch. Farben Z., 30, 392–454.
- 224 DIN 55 988 (1981) Bestimmung der Transparenz (Lasur) von pigmentierten und unpigmentierten Systemen; Farbmetrisches Verfahren.
- 225 Maikowski, M. (1971) Farbe+Lack, 77, 640–647.
- 226 Manufacturer: Original Hanau Quarzlampen GmbH, Hanau.
- 227 Hauser, P., Herrmann, H., and Honigmann, B. (1970) Farbe+Lack, 76, 545–550; Hauser, P., Herrmann, H. and Honigmann, B. (1971) Farbe+Lack, 77, 1097–1106.
- 228 Herbst, W. and Merkle, K. (1968) Farbe+Lack, 74, 1072–1089, 1174–1179.
- 229 Carr, W.C. (1967) J. Oil Color Chem. Assoc., 50, 1115–1155.
- 230 Gall, L. and Kaluza, U. (1975) DEFAZET Dtsch. Farben Z., 29, 102–115.
- 231 Kaluza, U. (1979) DEFAZET Dtsch. Farben Z., 33, 355–359, 399–411.
- 232 Crowl, V.T. (1972) J. Oil Color Chem. Assoc., 55, 388; Fürbeth, W. Grundmeier, G. and Stratmann, M. (1996) Farbe+Lack, 102, 78–84.
- 233 Haselmeyer, F. (1968) Farbe+Lack, 74, 662–668.
- 234 Herbst, W. and Merkle, K. (1969) Farbe+Lack, 75, 1137–1157.
- 235 DIN ISO 8780–2 (1990) Dispergieren mit einer Schüttelmaschine. Dispersion using an oscillatory shaking machine.
- 236 Ostwald, W. (1900) Z. Phys. Chem., 34, 295.
- 237 Noyes, A. and Whitney, W. (1897) Z. Phys. Chem., 23, 289.
- 238 DIN 54 002 (1982) Herstellung und Handhabung des Graumaßstabes zur Bewertung des Ausblutens. ISO 105–A03–1987: Textiles – Test for color fastness, Part A03: Grey scale for assessing staining.
- 239 Fettis, G. (1994) Automotive Paints and Coatings, VCH Verlagsgesellschaft, Weinheim.
- 240 Goldschmidt, A. (1978) Farbe+Lack, 84, 675–680.
- 241 Herbst, W. and Hafner, O. (1976) Farbe + Lack, 82, 393–411.
- 242 Beeferman, H.L. and Water-Bone, J. Coatings, (1978) 1, 4.
- 243 Dören, K., Freitag, W., and Stoye, D. (1993) Wasserlacke: Umweltschonende Alternative für Beschichtungen. Grundlagen, Rohstoffe, Entwicklungen, Technologien, 2nd edn, Verlag TÜV Rheinland, Köln.
- 244 Burckhard, W. and Luthardt, H.J. (1979) J. Oil Colour Chem. Assoc., 62, 375–385.
- 245 European Council Directive 1999/13/EC of March 1999; Commission Decision 2000/541/EC.
- 246 Rybny, C.B. et al. (1974) J. Paint Technol., 46 (596), 60–69.
- 247 T.A. Du Plessis and De Hollain, (1979) J. Oil Colour Chem. Assoc., 62 239–245; Pappas, P.S. (1980) in Sixth International Conference in Organic Coatings Science and Technology: Proceeding, presented in Athens, Greece 1980 (ed. G.D. Parfitt and A.V. Patsis), Advances in Organic Coatings Science and Technology, Technomic Pub. Co., Westport, CT, pp. 587–597.
- 248 Schäfer, H. and Wilker, G. Hoechst A.G., personal communication.
- 249 Schlusen, W. (1993) Farbe+Lack, 99, 778–781.
- 250 Meyer, B.D. (1979) in Fifth International Conference in Organic Coatings Science and Technology: Proceeding, presented in Athens, Greece 1979 (ed. G.D. Parfitt and A.V. Patsis), Advances in Organic Coatings Science and Technology, Technomic Pub. Co., Westport, CT pp. 177–206.
- 251 Schäfer, H. and Wallisch, G. (1981) J. Oil Color Chem. Assoc., 64, 405–414.
- 252 Kaluza, U. (1979) DEFAZET Dtsch. Farben Z., 33, 399–411.
- 253 Haselmeyer, F. (1968) Farbe+Lack, 74, 662–668.
- 254 Kresse, P. (1970) DEFAZET Dtsch. Farben Z., 24, 521–526.
- 255 Percy, E.J. and Nouwens, F. (1979) J. Oil Color Chem. Assoc., 62, 392–400.
- 256 Brushwell, W. (1974) Farbe+Lack, 80, 639–642; Brushwell, W. (1979) Farbe+Lack, 85, 1035–1038.
- 257 Newton, D.S. (1973) J. Oil Color Chem. Assoc., 56, 566–575.
- 258 Schmid, E.V. (1979) Farbe+Lack, 85, 744–748.
- 259 Gutbrod, R. (1963) Farbe+Lack, 69, 889–892.
- 260 prEN 13900-5, Pigments and extenders — Methods of dispersion and assessment of dispersion characteristics in plastics — Part 5: Determination by filter pressure rise test.
- 261 Richter, J. (XXXX) Das Einfärben von Kunststoffen und Kunstfasern in der Masse, Hoechst, SD 119.
- 262 DIN 53235/2 (1974) Test on specimens having standard depth of shade, adjustment of specimens to standard depth of shade.
- 263 DIN 53 774 T 1 (1986) also DIN 53 775 T 1, Prüfung von Pigmenten in PVC hart bzw. PVC weich; Zusammensetzung und Herstellen der Grundmischungen.
- 264 DIN 53 773, Prüfung von Farbmitteln in Polyvinylchlorid-Pasten (Plastisolen).
- 265 DIN 53 774 T 2 (1986) also DIN 53 775 T 2, Herstellen der Probekörper.
- 266 DIN 53 775 T 6 (1982) Bestimmung der Hitzebeständigkeit im Wärmeschrank.
- 267 DIN 53 775 T 3 (1984) Bestimmung des Ausblutens.
- 268 DIN 53 483, Bestimmung der dielektrischen Eigenschaften.
- 269 DIN 47 002 (1987) BS 6746, Farben und Farbkurzzeichen für Kabel und isolierte Leitungen.
- 270 Lenz, H.-J. (1976) Kunststoffe, 66, 683–687.
- 271 Kuckelsberg, F. (1972) Kunstst. Rundsch., 654–660.
- 272 Teige, W.K.J. (1975) Society of Plastic Engineers, 33rd Annual Technical Conference: Atlanta, Georgia 5-8 May 1975, Society of Plastic Engineers, pp. 57–59.
- 273 Teige, W.K.J. (1983) Chemiefasern/Textilind., 33/85, 636–642.
- 274 Welfers, E. (1976) Chemiefasern/Textilind., 26/78, 1079–1086; 27/79 (1977) 42–59.
- 275 Hoechst AG (1976) DE-AS 2 608 481.
- 276 Steinlin, F. and Wampetich, M.J. (1980) Melliand Textilber., 61, 509–513.
- 277 Teige, W.K.J. (1983) Chemiefasern/Textilind., 33/85, 127–131.
- 278 Teige, W.K.J. (1981) Chemiefasern/Textilind., 31/83, 632–636.
- 279 Steier, J. Das Färben von Holz, Hoechst AG, 6.76, SD 441.
- 280 Farbstoffe und Pigmentpräparationen für die Papierindustrie. Hoe 3023.
- 281 Techn. Rat aus Hoechst Pigment (1975) Papieroberflächenfärbung in der Leimpresse, 30.
- 282 Techn. Rat aus Hoechst (1969) Das Färben von Seifen, 21.
- 283 Schmidt, M.U. and Kliemt, K. (2015) German Patent Application DE 102013114793.3.