
The Kleisli Query System as a Backbone for Bioinformatics Data Integration and Analysis
Biological data is characterized by a wide range of data types from the plain text of laboratory records and literature, to nucleic acid and amino acid sequences, 3D structures of molecules, high-resolution images of cells and tissues, diagrams of biochemical pathways and regulatory networks, to various experimental outputs from technologies as diverse as microarrays, gels, and mass spectrometry. These data are stored in a large number of databases across the Internet. In addition to online interfaces for querying and searching the underlying repository data, many Web sites also provide specific computational tools or programs for analysis of data. In this chapter the term data sources is used loosely to refer to both databases and computational analysis tools.
Until recently, data sources were set up as autonomous Web sites by individual institutions or research laboratories. Data sources vary considerably in contents, access methods, capacity, query processing, and services. The major difficulty is that the data elements in various public and private data sources are stored in extremely heterogeneous formats and database management systems that are often ad hoc, application-specific, or vendor-specific. For example, scientific literature, patents, images, and other free-text documents are commonly stored in unstructured formats like plain text files, hypertext markup language (HTML) files, and binary files. Genomic, microarray gene expression, or proteomic data are routinely stored in Excel spreadsheets, semi-structured extensible markup language (XML), or structured relational databases like Oracle, Sybase, DB2, and Informix. The National Center for Biotechnology Information (NCBI) in Bethesda, Maryland, which is the largest repository for genetic information, supplies GenBank reports and GenPept reports in HTML format with an underlying highly nested data model based on ASN.1 [1]. The computational analysis tools or applications suffer from a similar scenario: They require specific input and output data formats. The output of one program is not immediately compatible with the input requirement of other programs. For example, the most popular Basic Local Alignment Search Tool (BLAST) database search tool requires a specific format called FASTA for sequence input.
In addition to data format variations, both the data content and data schemas of these databases are constantly changing in response to rapid advances in research and technology. As the amount of data and databases continues to grow on the Internet, it generates another bottleneck in information integration at the semantic level. There is a general lack of standards in controlled vocabulary for consistent naming of biomedical terms, functions, and processes within and between databases. In naming genes and proteins alone, there is much confusion. For example, a simple transcription factor, the CCAAT/enhancer-binding protein beta, is referred to by more than a dozen names in the public databases, including CEBPB, CRP2, and IL6DPB.
For research and discovery, the biologist needs access to up-to-date data and best-of-breed computational tools for data analyses. To achieve this goal, the ability to query across multiple data sources is not enough. It also demands the means to transform and transport data through various computational steps seamlessly. For example, to investigate the structure and function of a new protein, users must integrate information derived from sequence, structure, protein domain prediction, and literature data sources. Should the steps to prepare the data sets between the output of one step to the input of the next step have to be carried out manually, which requires some level of programming work (such as writing Perl scripts), the process would be very inefficient and slow.
In short, many bioinformatics problems require access to data sources that are large, highly heterogeneous and complex, constantly evolving, and geographically dispersed. Solutions to these problems usually involve many steps and require information to be passed smoothly and usually to be transformed between the steps. The Kleisli system is designed to handle these requirements directly by providing a high-level query language, simplified SQL (sSQL), that can be used to express complicated transformations across multiple data sources in a clear and simple way.1
The design and implementation of the Kleisli system are heavily influenced by functional programming research, as well as database query language research. Kleisli’s high-level query language, sSQL, can be considered a functional programming language2 that has a built-in notion of bulk data types3 suitable for database programming and has many built-in operations required for modern bioinformatics. Kleisli is implemented on top of the functional programming language Standard ML of New Jersey (SML). Even the data format Kleisli uses to exchange information with the external world is derived from ideas in type inference in functional programming languages.
This chapter provides a description of the Kleisli system and a discussion of various aspects of the system, such as data representation, query capability, optimizations, and user interfaces. The materials are organized as follows: Section 6.1 introduces Kleisli with a well-known example. Section 6.2 presents an overview of the Kleisli system. Section 6.3 discusses the data model, data representation, and exchange format of Kleisli. Section 6.4 gives more example queries in Kleisli and comments on the expressive power of its core query language. Section 6.5 illustrates Kleisli’s ability to use flat relational databases to store complex objects transparently. Section 6.6 lists the kind of data sources supported by the Kleisli system and shows the ease of implementing wrappers for Kleisli. Section 6.7 gives an overview of the various types of optimizations performed by the Kleisli query optimizer. Section 6.8 describes both the Open Database Connectivity (ODBC)-or Java Database Connectivity (JDBC)-like programming interfaces to Kleisli in Perl and Java, as well as its Discovery Builder graphical user interface. Section 6.9 contains a brief survey of other well-known proposals for bioinformatics data integration.
6.1 MOTIVATING EXAMPLE
Before discussing the guts of the Kleisli system, the very first bioinformatics data integration problem solved using Kleisli is presented in Example 6.1.1. The query was implemented in Kleisli in 1994 [5] and solved one of the so-called “impossible” queries of a U.S. Department of Energy Bioinformatics Summit Report published in 1993 [6].
Example 6.1.1: The query was to “find for each gene located on a particular cytogenetic band of a particular human chromosome as many of its non-human homologs as possible.” Basically, the query means “for each gene in a particular position in the human genome, find dioxyribonucleic acid (DNA) sequences from non-human organisms that are similar to it.”
In 1994, the main database containing cytogenetic band information was the Genome Database (GDB) [7], which was a Sybase relational database. To find homologs, the actual DNA sequences were needed, and the ability to compare them was also needed. Unfortunately, that database did not keep actual DNA sequences. The actual DNA sequences were kept in another database called GenBank [8]. At the time, access to GenBank was provided through the ASN.1 version of Entrez [9], which was at the time an extremely complicated retrieval system. Entrez also kept precomputed homologs of GenBank sequences.
So, the evaluation of this query needed the integration of GDB (a relational database located in Baltimore, Maryland) and Entrez (a non-relational database located in Bethesda, Maryland). The query first extracted the names of genes on the desired cytogenetic band from GDB, then accessed Entrez for homologs of these genes. Finally, these homologs were filtered to retain the non-human ones. This query was considered “impossible” as there was at that time no system that could work across the bioinformatics sources involved due to their heterogeneity, complexity, and geographical locations. Given the complexity of this query, the sSQL solution below is remarkably short.

The first three lines connect to GDB and map two tables in GDB to Kleisli. After that, these two tables could be referenced within Kleisli as if they were two locally defined sets, locus and eref. The next few lines extract from these tables the accession numbers of genes on Chromosome 22, use the Entrez function naget-homolog-summary to obtain their homologs, and filter these homologs for non-human ones. Notice that the from-part of the outer select-construct is of the form {select u …} H. This means that H is the entire set returned by select u …, thus allowing to manipulate and return all the non-human homologs as a single set H.
Besides the obvious smoothness of integration of the two data sources, this query is also remarkably efficient. On the surface, it seems to fetch the locus table in its entirety once and the eref table in its entirety n times from GDB (a naive evaluation of the comprehension would be two nested loops iterating over these two tables). Fortunately, in reality, the Kleisli optimizer is able to migrate the join, selection, and projections on these two tables into a single efficient access to GDB using the optimizing rules from a later section of this chapter. Furthermore, the accesses to Entrez are also automatically made concurrent.
Since this query, Kleisli and its components have been used in a number of bioinformatics projects such as GAIA at the University of Pennsylvania,4 Transparent Access to Multiple Bioinformatics Information Sources (TAMBIS) at the University of Manchester [11, 12], and FIMM at Kent Ridge Digital Labs [13]. It has also been used in constructing databases by pharmaceutical/biotechnology companies such as SmithKline Beecham, Schering-Plough, GlaxoWellcome, Genomics Collaborative, and Signature Biosciences. Kleisli is also the backbone of the Discovery Hub product of geneticXchange Inc.5
6.2 APPROACH
The approach taken by the Kleisli system is illustrated by Figure 6.1. It is positioned as a mediator system encompassing a complex object data model, a high-level query language, and a powerful query optimizer. It runs on top of a large number of lightweight wrappers for accessing various data sources. There is also a number of application programming interfaces that allow Kleisli to be accessed in an ODBC-or JDBC-like fashion in various programming languages for a various applications.

The Kleisli system is extensible in several ways. It can be used to support several different high-level query languages by replacing its high-level query language module. Currently, Kleisli supports a comprehension syntax-based language called CPL [2, 14, 15] and a nested relationalized version of SQL called sSQL. Only sSQL is used throughout this chapter. The Kleisli system can also be used to support many different types of external data sources by adding new wrappers, which forward Kleisli’s requests to these sources and translate their replies into Kleisli’s exchange format. These wrappers are lightweight and new wrappers are generally easy to develop and insert into the Kleisli system. The optimizer of the Kleisli system can also be customized by different rules and strategies.
When a query is submitted to Kleisli, it is first processed by the high-level query language module, which translates it into an equivalent expression in the abstract calculus Nested Relational Calculus (NRC). NRC is based on the calculus described in Buneman’s “Principles of Programming with Complex Objects and Collection Types” [16] and was chosen as the internal query representation because it is easy to manipulate and amenable to machine analysis. The NRC expression is then analyzed to infer the most general valid type for the expression and is passed to the query optimizer. Once optimized, the NRC expression is then compiled into calls to a library of routines for complex objects underlying the complex object data model. The resulting compiled code is then executed, accessing drivers and external primitives as needed through pipes or shared memory. Each of these components is considered in further detail in the next several sections.
6.3 DATA MODEL AND REPRESENTATION
The data model, data representation, and data exchange format of the Kleisli system are presented in this section. The data model underlying the Kleisli system is a complex object type system that goes beyond the sets of atomic records or flat relations type systems of relational databases [17]. It allows arbitrarily nested records, sets, lists, bags, and variants. A variant is also called a tagged union type, and it represents a type that is either this or that. The collection or bulk types—sets, bags, and lists—are homogeneous. To mix objects of different types in a set, bag, or list, it is necessary to inject these objects into a variant type.
In a relational database, the sole bulk data type is the set. Furthermore, this set is only allowed to contain records where each field is allowed to contain an atomic object such as a number or a string. Having such a restricted bulk data type presents at least two problems in real-life applications. First, the particular bulk data type may not be a natural model of real data. Second, the particular bulk data type may not be an efficient model of real data. For example, when restricted to the flat relational data model, the GenPept report in Example 6.3.1 must be split into many separate tables to be stored in a relational database without loss. The resulting multi-table representation of the GenPept report is conceptually unnatural and operationally inefficient. A person querying the resulting data must pay the mental overhead of understanding both the original GenPept report and its badly fragmented multi-table representation. The user may also have to pay the performance overhead of having to reassemble the original GenPept report from its fragmented multi-table representation to answer queries. As another example, limited with the set type only, even if nesting of sets is allowed, one may not be able to model MEDLINE reports naturally. A MEDLINE report records information on a published paper, such as its title and its authors. The order in which the authors are listed is important. With only sets, one must pair each author explicitly with a number representing his or her position in order of appearance. Whereas with the data type list, this cumbersome explicit pairing with position becomes unnecessary.
Example 6.3.1: The GenPept report is the format chosen by NCBI to represent information on amino acid sequence. While an amino acid sequence is a string of characters, certain regions and positions of the string, such as binding sites and domains, are of special biological interest. The feature table of a GenPept report is the part of the GenPept report that documents the positions of these regions of special biological interest, as well as annotations or comments on these regions. The following type represents the feature table of a GenPept report from Entrez [9].

It is an interesting type because one of its fields (#feature) is a set of records, one of whose fields (#anno) is in turn a list of records. More precisely, it is a record with four fields #uid, #title, #accession, and #feature. The first three of these store values of types num, string, and string respectively. The #uid field uniquely identifies the GenPept report. The #feature field is a set of records, which together form the feature table of the corresponding GenPept report. Each of these records has four fields: #name, #start, #end, and #anno. The first three of these have types string, num, and num respectively. They represent the name, start position, and end position of a particular feature in the feature table. The #anno field is a list of records. Each of these records has two fields #anno_name and #descr, both of type string. These records together represent all annotations on the corresponding feature.
In general, the types are freely formed by the syntax:
![]()
Here num, string, and bool are the base types. The other types are constructors and build new types from existing types. The types {t}, {|t|}, and [t] respectively construct set, bag, and list types from type t. The type (l1 : t1, …, ln : tn) constructs record types from types t1, …, tn. The type <l1 : t1, …, ln : tn> constructs variant types from types t1, …, tn. The flat relations of relational databases are basically sets of records, where each field of the record is a base type; in other words, relational databases have no bags, no lists, no variants, no nested sets, and no nested records. Values of these types can be represented explicitly and exchanged as follows, assuming that the instances of e are values of appropriate types: (l1 : e1, …, ln : en) for records; <l : e> for variants; {e1, …, en} for sets; {|e1, …, en|} for bags; and [e1, …, en] for lists.
Example 6.3.2: Part of the feature table of GenPept report 131470, a tyrosine phosphatase 1C sequence, is shown in the following.

The particular feature goes from amino acid 0 to amino acid 594, which is actually the entire sequence, and has two annotations: The first annotation indicates that this amino acid sequence is derived from a mouse DNA sequence. The second is a cross reference to the NCBI taxonomy database.
The schemas and structures of all popular bioinformatics databases, flat files, and software are easily mapped into this data model. At the high end of data structure complexity are Entrez [9] and AceDB [18], which contain deeply nested mixtures of sets, bags, lists, records, and variants. At the low end of data structure complexity are the relational database systems [17] such as Sybase and Oracle, which contain flat sets of records. Currently, Kleisli gives access to more than 60 of these and other bioinformatics sources. The reason for this ease of mapping bioinformatics sources to Kleisli’s data model is that they are all inherently composed of combinations of sets, bags, lists, records, and variants. Kleisli’s data model directly and naturally maps sets to sets, bags to bags, lists to lists, records to records, and variants to variants without having to make any (type) declaration beforehand.
The last point deserves further consideration. In a dynamic, heterogeneous environment such as that of bioinformatics, many different database and software systems are used. They often do not have anything that can be thought of as an explicit database schema. Further compounding the problem is that research biologists demand flexible access and queries in ad hoc combinations. Thus, a query system that aims to be a general integration mechanism in such an environment must satisfy four conditions. First, it must not count on the availability of schemas. It must be able to compile any query submitted based solely on the structure of that query. Second, it must have a data model that the external database and software systems can easily translate to without doing a lot of type declarations. Third, it must shield existing queries from evolution of the external sources as much as possible. For example, an extra field appearing in an external database table must not necessitate the recompilation or rewriting of existing queries over that data source. Fourth, it must have a data exchange format that is straightforward to use so it does not demand too much programming effort or contortion to capture the variety of structures of output from external databases and software.
Three of these requirements are addressed by features of sSQL’s type system. sSQL has polymorphic record types that allow to express queries such as:
![]()
which defines a function that returns names of people in R earning more than $1000. This function is applicable to any R that has at least the name and the salary fields, thus allowing the input source some freedom to evolve.
In addition, sSQL does not require any type to be declared at all. The type and meaning of any sSQL program can always be completely inferred from its structure without the use of any schema or type declaration. This makes it possible to plug in any data source logically without doing any form of schema declaration, at a small acceptable risk of run-time errors if the inferred type and the actual structure are not compatible. This is an important feature because most biological data sources do not have explicit schemas, while a few have extremely large schemas that take many pages to write down—for example, the ASN.l schema of Entrez [1]—making it impractical to have any form of declaration.
As for the fourth requirement, a data exchange format is an agreement on how to lay out data in a data stream or message when the data is exchanged between two systems. In this context, it is the format for exchanging data between Kleisli and all the bioinformatics sources. The data exchange format of Kleisli corresponds one-to-one with Kleisli’s data model. It provides for records, variants, sets, bags, and lists; and it allows these data types to be composed freely. In fact, the data exchange format completely adopts the syntax of the data representation described earlier and illustrated in Example 6.3.2. This representation has the interesting property of not generating ambiguity. For instance, a set symbol { represents a set, whereas a parenthesis (denotes a record. In short, this data exchange format is self describing. The basic specification of the data exchange format of Kleisli is summarized in Figure 6.2. For a more detailed account, please see Wong’s paper on Kleisli from the 2000 IEEE Symposium on Bioinformatics and Bio-engineering [19].
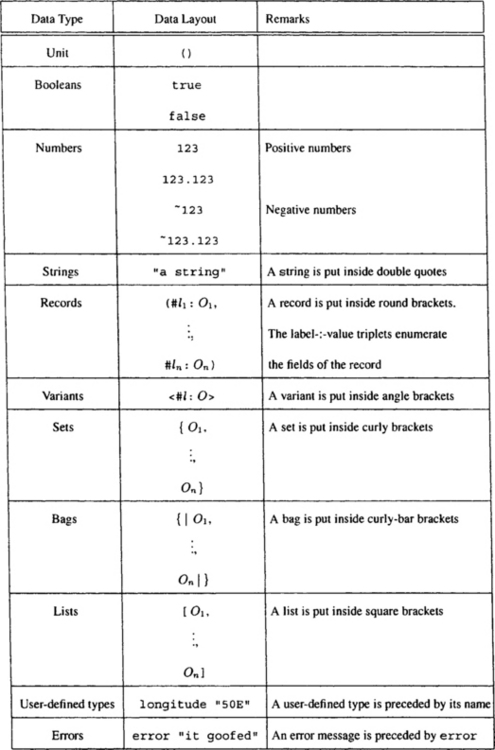
FIGURE 6.2 The basic form of the Kleisli Exchange Format. Punctuations and indentations are not significant. The semicolon indicates the end of a complex object. Multiple complex objects can be laid out in the same stream.
A self-describing exchange format is one in which there is no need to define in advance the structure of the objects being exchanged. That is, there is no fixed schema and no type declaration. In a sense, each object being exchanged carries its own description. A self-describing format has the important property that, no matter how complex the object being exchanged is, it can be easily parsed and reconstructed without any schema information. The ISO ASN.1 standard [20] on open systems interconnection explains this advantage. The schema that describes its structure needs to be parsed before ASN.1 objects, making it necessary to write two complicated parsers instead of one simple parser.
6.4 QUERY CAPABILITY
sSQL is the primary query language of Kleisli used in this chapter. It is based on the de facto commercial database query language SQL, except for extensions made to cater to the nested relational model and the federated heterogeneous data sources. Rather than giving the complete syntax, sSQL is illustrated with a few examples on a set of feature tables DB.
Example 6.4.1: The query below “extracts the titles and features of those elements of a data source DB whose titles contain ‘tyrosine’ as a substring.”

This query is a simple project-select query. A project-select query is a query that operates on one (flat) relation or set. Thus, the transformation such a query can perform is limited to selecting some elements of the relation and extracting or projecting some fields from these elements. Except for the fact that the source data and the result may not be in first normal form, these queries can be expressed in a relational query language. However, sSQL can perform more complex restructurings such as nesting and unnesting not found in SQL, as shown in the following examples.
Example 6.4.2: The following query flattens the source DB completely. 12s is a function that converts a list into a set.

The next query demonstrates how to express nesting in sSQL. Notice that the entries field is a complex object having the same type as DB.


The next couple of more substantial queries are inspired by one of the most advanced functionalities of the EnsMart interface of the EnsEMBL system [21].
Example 6.4.3: The feature table of a GenBank report has the type below. The field #position of a feature entry is a list indicating the start and stop positions of that feature. If the feature entry is a CDS, this list corresponds to the list of exons of the CDS. The field #anno is a list of annotations associated with the feature entry.

Given a set DB of feature tables of GenBank chromosome sequences, one can extract the 500 bases up stream of the translation initiation sites of all disease genes—in the sense that these genes have a cross reference to the Online Mendelian Inheritance of Man database (OMIM)—on the positive strand in DB as below.

Similarly, one can extract the first exons of these same genes as follows:
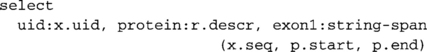

These two example queries illustrate how a high-level query language makes it possible to extract very specific output in a relatively straightforward manner.
The next query illustrates a more ambitious example of an in silico discovery kit (ISDK). Such a kit prescribes experimental steps carried out in computers very much like the experimental protocol carried out in wet-laboratories for a specific scientific investigation. From the perspective of Kleisli, an in silico discovery kit is just a script written in sSQL, and it performs a defined information integration task very similar to an integrated electronic circuit. It takes an input data set and parameters from the user, executes and integrates the necessary computational steps of database queries and applications of analysis programs or algorithms, and outputs a set of results for specific scientific inquiry.
Example 6.4.4: The simple in silico discovery kit illustrated in Figure 6.3 demonstrates how to use an available ontology data source to get around the problem of inconsistent naming in genes and proteins and to integrate information across multiple data sources. It is implemented in the following sSQL script.6 With the user input of gene name G, the ISDK performs the following tasks: “First, it retrieves a list of aliases for G from the gene nomenclature database provided by the Human Genome Organization (HUGO). Then it retrieves information for diseases associated with this particular protein in OMIM, and finally it retrieves all relevant references from MEDLINE.”

FIGURE 6.3 An “in silico discovery kit” that uses an available ontology data source to get around the problem of inconsistent naming in genes and proteins, and integrates information across multiple data sources.


For instance, this query get-info-by-genename can be invoked with the transcription factor CEBPB as input to obtain the following result.


Such queries fulfill many of the requirements for efficient in silico discovery processes: (1) Their modular nature gives scientists the flexibility to select and combine specific queries for specific research projects; (2) they can be executed automatically by Kleisli in batch mode and can handle large data volumes; (3) their scripts are re-usable to perform repetitive tasks and can be shared among scientific collaborators; (4) they form a base set of templates that can be readily modified and refined to meet different specifications and to make new queries; and (5) new databases and new computational tools can be readily incorporated to existing scripts.
The flexibility and power shown in these sSQL examples can also be experienced in Object-Protocol Model (OPM) [22] and to a lesser extent in DiscoveryLink [23]. With good planning, a specialized data integration system can also achieve great flexibility and power within a more narrow context. For example, the EnsMart tool of EnsEMBL [21] is a well-designed interface that helps a non-programmer build complex queries in a simple way. In fact, an equivalent query to the first sSQL query in Example 6.4.3 can be also be specified using EnsMart with a few clicks of the mouse. Nevertheless, there are some unanticipated cases that cannot be expressed in EnsMart, such as the second sSQL query in Example 6.4.3.
While the syntactic basis for sSQL is SQL, its theoretical inspiration came from a paper by Tannen, Buneman, and Nagri [24] where structural recursion was presented as a query language. However, structural recursion presents two difficulties. The first is that not every syntactically correct structural recursion program is logically well defined [25]. The second is that structural recursion has too much expressive power because it can express queries that require exponential time and space.
In the context of databases, which are typically very large, programs (queries) are usually restricted to those that are practical in the sense that they are in a low complexity class such as LOGSPACE, PTIME, or TC0. In fact, one may even want to prevent any query that has greater than O(n × log n) complexity, unless one is confident that the query optimizer has a high probability of optimizing the query to no more than O(n × log n) complexity. Database query languages such as SQL, therefore, are designed in such a way that joins are easily recognized because joins are the only operations in a typical database query language that require O(n2) complexity if evaluated naively.
Thus, Tannen and Buneman suggested a natural restriction on structural recursion to reduce its expressive power and to guarantee it is well defined. Their restriction cuts structural recursion down to homomorphisms on the commutative idempotent monoid of sets, revealing a telling correspondence to monads [15]. A nested relational calculus, which is denoted here by NRC, was then designed around this restriction [16]. NRC is essentially the simply typed lambda calculus extended by a construct for building records, a construct for decomposing records by field selection, a construct for building sets, and a construct for decomposing sets by means of the restriction on structural recursion. Specifically, the construct for decomposing sets is ∪ which forms a set by taking the big union of e1 [o/x] over each o in the set e2.
The expressive power of NRC and its extensions are studied in numerous studies [16, 26–29]. Specifically, the NRC core has exactly the same power as all the standard nested relational calculi and when restricted to flat tables as input-output, it has exactly the same power as the relational calculus. In the presence of arithmetics and a summation operator, when restricted to flat tables as input-output, it has exactly the power of entry-level SQL. Furthermore, it captures standard nested relational queries in a high-level manner that is easy for automated optimizer analysis. It is also easy to translate a more user-friendly surface syntax, such as the comprehension syntax or the SQL select-from-where syntax, into this core while allowing for full-fledged recursion and other operators to be imported easily as needed into the system.
6.5 WAREHOUSING CAPABILITY
Besides the ability to query, assemble, and transform data from remote heterogeneous sources, it is also important to be able to conveniently warehouse the data locally. The reasons to create local warehouses are several: (1) It increases efficiency; (2) it increases availability; (3) it reduces the risk of unintended denial of service attacks on the original sources; and (4) it allows more careful data cleansing that cannot be done on the fly. The warehouse should be efficient to query and easy to update. Equally important in the biology arena, the warehouse should model the data in a conceptually natural form. Although a relational database system is efficient for querying and easy to update, its native data model of flat tables forces an unnatural and unnecessary fragmentation of data to fit third normal form.
Kleisli does not have its own native database management system. Instead, Kleisli has the ability to turn many kinds of database systems into an updatable store conforming to its complex object data model. In particular, Kleisli can use flat relational database management systems such as Sybase, Oracle, and MySQL, to be its updatable complex object store. It can even use these systems simultaneously. This power of Kleisli is illustrated using the example of GenPept reports.
Example 6.5.1: Create a warehouse of GenPept reports and initialize it to reports on protein tyrosine phosphatases. Kleisli provides several functions to access GenPept reports remotely from Entrez [9]. One of them is aa-get-seqfeat-general, which retrieves GenPept reports matching a search string.

In this example, a table genpept is created in the local Oracle database system. This table has two columns, uid for recording the unique identifier and detail for recording the GenPept report. A LONG data type is used for the detail column of this table. However, recall from Example 6.3.2 that each GenPept report is a highly nested complex object. There is therefore a mismatch between LONG (which is essentially a big, uninterpreted string) and the complex structure of a GenPept report. This mismatch is resolved by the Kleisli system, which automatically performs the appropriate encoding and decoding. Thus, as far as the Kleisli user is concerned, x.detail has the type of GenPept report as given in Example 6.3.1. So the user can ask for the title of a report as straightforwardly as x. detail. title. Note that encoding and decoding are performed to map the complex object transparently into the space provided in the detail column; that is, the Kleisli system does not fragment the complex object to force it into third normal form.
There are two possible techniques to use a flat relational database system as a nested relational store. This first is to add a layer on top of the underlying flat relational database system to perform automatic normalization of nested relations into the third normal form. This is the approach taken by systems such as OPM [22]. Such an approach may lead to performance problems as the database system may be forced to perform many extra joins under certain situations. The second technique is to add a layer on top of the underlying flat relational database system to perform automatic encoding and decoding of nested components into long strings. This is the technique adopted in Kleisli because it avoids unnecessary joins and because it is a simple extension—without significant additional overhead—to the handling of Kleisli’s data exchange format.
6.6 DATA SOURCES
The standard version of the Kleisli system marketed by geneticXchange, Inc. supports more than 60 types of data sources. These include the categories below.
![]() Relational database management systems: All popular relational database management systems are supported, such as Oracle, Sybase, DB2, Informix, and MySQL. The support for these systems is quite sophisticated. For example, the previous section illustrates how the Kleisli system can turn these flat database systems transparently into efficient complex object stores that support both read and write access. A later section also shows that the Kleisli system is able to perform significant query optimization involving these systems.
Relational database management systems: All popular relational database management systems are supported, such as Oracle, Sybase, DB2, Informix, and MySQL. The support for these systems is quite sophisticated. For example, the previous section illustrates how the Kleisli system can turn these flat database systems transparently into efficient complex object stores that support both read and write access. A later section also shows that the Kleisli system is able to perform significant query optimization involving these systems.
![]() Bioinformatics analysis packages: Most popular packages for analysis of protein sequences and other biological data are supported. These packages include both Web-based and/or locally installed versions of WU-BLAST [30], Gapped BLAST [31], FASTA, CLUSTAL W [32], HMMER, BLOCKS, Profile Scan (PFSCAN) [33], NNPREDICT, PSORT, and many others.
Bioinformatics analysis packages: Most popular packages for analysis of protein sequences and other biological data are supported. These packages include both Web-based and/or locally installed versions of WU-BLAST [30], Gapped BLAST [31], FASTA, CLUSTAL W [32], HMMER, BLOCKS, Profile Scan (PFSCAN) [33], NNPREDICT, PSORT, and many others.
![]() Biological databases: Many popular data sources of biological information are also supported by the Kleisli system, including AceDB [18], Entrez [9], LocusLink, UniGene, dbSNP, OMIM, PDB, SCOP [34], TIGR, KEGG, and MEDLINE. For each of these sources, Kleisli typically provides many access functions corresponding to different capabilities of the sources. For example, Kleisli provides about 70 different but systematically organized functions to access and extract information from Entrez.
Biological databases: Many popular data sources of biological information are also supported by the Kleisli system, including AceDB [18], Entrez [9], LocusLink, UniGene, dbSNP, OMIM, PDB, SCOP [34], TIGR, KEGG, and MEDLINE. For each of these sources, Kleisli typically provides many access functions corresponding to different capabilities of the sources. For example, Kleisli provides about 70 different but systematically organized functions to access and extract information from Entrez.
![]() Patent databases: Currently only access to the United States Patent and Trademark Office (USPTO) is supported.
Patent databases: Currently only access to the United States Patent and Trademark Office (USPTO) is supported.
![]() Interfaces: The Kleisli system also provides means for parsing input and writing output in HTML and XML formats. In addition, programming libraries are provided for Java and Perl to interface directly to Kleisli in a fashion similar to JDBC and ODBC. A graphical user interface called Discovery Builder is also available.
Interfaces: The Kleisli system also provides means for parsing input and writing output in HTML and XML formats. In addition, programming libraries are provided for Java and Perl to interface directly to Kleisli in a fashion similar to JDBC and ODBC. A graphical user interface called Discovery Builder is also available.
It is generally easy to develop a wrapper for a new data source, or modify an existing one, and insert it into Kleisli. There is no impedance mismatch between the data model supported by Kleisli and the data model necessary to capture the data source. The wrapper is therefore often a very lightweight parser that simply parses records in the data source and prints them out in Kleisli’s simple data exchange format.
Example 6.6.1: Let us consider the webomim-get-detail function used in Example 6.4.4. It uses an OMIM identifier to access the OMIM database and returns a set of objects matching the identifier. The output is of type:
![]()
Note that this is a nested relation: It consists of a set of records, and each record has three fields that are also of set types, namely #gene_map_locus, #alternative_titles, and #allelic_variants. This type of output would definitely present a problem if it had to be sent to a system based on the flat relational model, as the information would have to be re-arranged in these three fields to be sent into separate tables.
Fortunately, such a nested structure can be mapped directly into Kleisli’s exchange format. The wrapper implementor would only need to parse each matching OMIM record and write it out in a format as illustrated in the following:


Instead of needing to create separate tables to keep the sets nested inside each record, the wrapper simply prints the appropriate set brackets { and } to enclose these sets. Kleisli will automatically deal with them as they were handed over by the wrapper. This kind of parsing and printing is extremely easy to implement. Figure 6.4 shows the relevant chunk of Perl codes in the OMIM wrapper implementing webomim-get-detail.

6.7 OPTIMIZATIONS
A feature that makes Oracle and Sybase much more productive to use than a raw file system is the availability of a high-level query language. Such a query language allows users to express their needs in a declarative, logical way. All low-level details such as opening files, handling disk blocks, using indices, decoding record and field boundaries, and so forth are hidden away and are automatically taken care of. However, there are two prices to pay, one direct and one indirect. The direct one is that if a high-level query is executed naively, the performance may be poor. The same high-level command often can be executed in several logically equivalent ways. However, which of these ways is more efficient often depends on the state of the data. A good optimizer can take the state of the data into consideration and pick the more efficient way to execute the high-level query. The indirect drawback is that because the query language is at a higher level, certain low-level details of programming are no longer expressible, even if these details are important to achieving better efficiency. However, a user who is less skilled in programming is now able to use the system. Such a user is not expected to produce always efficient programs. A good optimizer can transform inefficient programs into more efficient equivalent ones. Thus, a good optimizer is a key ingredient of a decent database system and of a general data integration system that supports ad hoc queries.
The Kleisli system has a fairly advanced query optimizer. The optimizations provided by this optimizer include (1) monadic optimizations that are derived from the equational theory of monads, such as vertical loop fusion; (2) context-sensitive optimizations, which are those equations that are true only in special contexts and that generally rely on certain long-range relationships between sub-expressions, such as the absorption of sub-expressions in the then-branch of an if – then - else construct that are equivalent to the condition of the construct; (3) relational optimizations, which are optimizations relating to relational database sources such as the migration of projections, selections, and joins to the external relational database management system; and (4) many other optimizations such as parallelism, code motion, and selective introduction of laziness.
6.7.1 Monadic Optimizations
The restricted form of structural recursion corresponds to the presentation of monads by Kleisli [15, 16] and is expressed by the combinator ![]() obeying the following three equations:
obeying the following three equations:

This combinator is at the heart of the NRC, the abstract representation of queries in the implementation of sSQL. It earns its central position in the Kleisli system because it offers tremendous practical and theoretical convenience. The direct correspondence in sSQL is: select y from R x, f(x) y. This combinator is a key operator in the library of complex object routines in Kleisli. All sSQL queries can be and are first translated into NRC via Wadler’s identities [15, 16].
The practical convenience of the ![]() combinator is best seen in query optimizations.
combinator is best seen in query optimizations.
A well-known optimization rule is vertical loop fusion [35], which corresponds to the physical notion of getting rid of intermediate data and the logical notion of quantifier elimination. Such an optimization on queries in the comprehension syntax can be expressed informally as {e | G1, …, Gn, x ∈ {e′ | H1, …, Hm}, J1, …, Jk} → {e[e′/x] | G1 …, Gn, H1, …, Hm, J1[e′/x], …, Jk[e’/x]}. Such a rule in comprehension form is simple to grasp: The intermediate set built by the comprehension {e′ | H1, …, Hm} is eliminated in favor of generating the x on the fly. In practice, the rule is quite messy to implement because the informal “…” denotes any number of generator-filters in a comprehension. An immediate implementation would involve a nasty traversal routine to skip over the non-applicable Gi to locate the applicable x ∈ {e′ | H1, …, Hm} and Ji. The effect of the ![]() combinator on the optimization rule for vertical loop fusion is dramatic. This optimization is now expressed as
combinator on the optimization rule for vertical loop fusion is dramatic. This optimization is now expressed as ![]() . The informal and troublesome no longer appears. Such a rule can be coded straightforwardly in almost any implementation language.
. The informal and troublesome no longer appears. Such a rule can be coded straightforwardly in almost any implementation language.
To illustrate this point more concretely, it is necessary to introduce some detail from the implementation of the Kleisli system. Recall from the introductory section that Kleisli is implemented on top of SML. The type SYN of SML objects that represent queries in Kleisli is declared as:

All SML objects that represent optimization rules in Kleisli are functions and have type RULE:
![]()
If an optimization rule r can be successfully applied to rewrite an expression e to an expression e′, then r(e) = SOME(e′). If it cannot be successfully applied, then r(e) = NONE.
Now the vertical loop fusion has a very simple implementation.

6.7.2 Context-Sensitive Optimizations
The Kleisli optimizer has an extensible number of phases. Each phase is associated with a rule base and a rule-application strategy. A large number of rule-application strategies are supported, such as BottomUpOnce, which applies rules to rewrite an expression tree from leaves to root in a single pass. By exploiting higher-order functions, these rule-application strategies can be decomposed into a traversal component common to all strategies and a simple control component special for each strategy. In short, higher-order functions can generate these strategies extremely simply, resulting in a small optimizer core. To give some ideas on how this is done, some SML code fragments from the optimizer module mentioned are presented on the following pages.
The traversal component is a higher-order function shared by all strategies:
![]()
Recall that SYN is the type of SML object that represents query expressions. The Decompose function accepts a rewrite rule r and a query expression Q. Then it applies r to all immediate subtrees of Q to rewrite these immediate subtrees. Note that it does not touch the root of Q and it does not traverse Q—it just non-recursively rewrites immediate subtrees using r. It is, therefore, very straightforward and can be expressed as follows:

A rule-application strategy S is a function having the following type:
![]()
The precise definition of the type RULEDB is not important at this point and is deferred until later. Such a function takes in a rule base R and a query expression Q and optimizes it to a new query expression Q′ by applying rules in R according to the strategy S.
Assume that Pick: RULEDB -> RULE is an SML function that takes a rule base R and a query expression Q and returns NONE if no rule is applicable, and SOME(Q′) if some rule in R can be applied to rewrite Q to Q′. Then the control components of all the strategies mentioned earlier can be generated easily.
Example 6.7.2: The BottomUpOnce strategy applies rules in a leaves-to-root pass. It tries to rewrite each node at most once as it moves toward the root of the query expression. Here is its control component:

The following class of rules requires the use of multiple rule-application strategies. The scope of rules like the vertical loop fusion in the previous section is over the entire query. In contrast, this class of rules has two parts. The inner part is context sensitive, and its scope is limited to certain components of the query. The outer part scopes over the entire query to identify contexts where the inner part can be applied. The two parts of the rule can be applied using completely different strategies.
A rule base RDB is represented in the system as an SML record of type:

The Rules field of RDB stores the list of rules in RDB together with their names. The Trace field of RDB stores a function f that is to be used for tracing the usage of the rules in RDB. The DoTrace field of RDB stores a flag to indicate whether tracing is to be done. If tracing is indicated, then whenever a rule of name N in RDB is applied successfully to transform a query Q to Q′, the trace function is invoked as f N Q Q′ to record a trace. Normally, this simply means a message like “Q is rewritten to Q′ using the rule N” is printed. However, the trace function f is allowed to carry out considerably more complicated activities.
It is possible to exploit trace functions to achieve sophisticated transformations in a simple way. An example is the rule that rewrites if e1 then … e1 … else e3 to if e1 then … true … else e3. The inner part of this rule rewrites e1 to true. The outer part of this rule identifies the context and scope of the inner part of this rule: limited to the then-branch. This example is very intuitive to a human being. In the then-branch of a conditional, all sub-expressions identical to the test predicate of the conditional must eventually evaluate to true. However, such a rule is not so straightforward to express to a machine. The informal “…” are again in the way. Fortunately, rules of this kind are straightforward to implement in Kleisli.
Example 6.7.3: The if-then-else absorption rule is expressed by the AbsorbThen rule below. The rule has three clauses. The first clause says the rule should not be applied to an IfThenElse whose test predicate is already a Boolean constant because it would lead to non-termination otherwise. The second clause says the rule should be applied to all other forms of IfThenElse. The third clause says the rule is not applicable in any other situation.

The second clause is the meat of the implementation. The inner part of the rewrite if e1 then … e1 … else e3 to if e1 then … true … else e3 is captured by the function Then, which rewrites any e identical to e1 to true. This function is then supplied as the rule to be applied using the TopDownOnce strategy within the scope of the then-branch … e1 … using the ContextSensitive rule generator given as follows.

This ContextSensitive rule generator is re-used in many other context-sensitive optimization rules, such as the rule for migrating projections to external relational database systems to be presented shortly.
6.7.3 Relational Optimizations
Relational database systems are the most powerful data sources to which Kleisli interfaces. These database systems are equipped with the ability to perform sophisticated transformations expressed in SQL. A good optimizer should aim to migrate as many operations in Kleisli to these systems as possible. There are four main optimizations that are useful in this context: the migration of projections, selections, and joins on a single database; and the migration of joins across two databases. The Kleisli optimizer has four different rules to exploit these four opportunities.
A special case of the rule for migrating P is to rewrite select x.name from (process “select * from T” using A) x to select x.name from (process “select name from T” using A) x, where process Q using A denotes sending an SQL query Q to a relational database A. In the original query, the entire table T has to be retrieved. In the rewritten query, only one column of that table has to be retrieved. More generally, if x is from a relational database system and every use of x is in the context of a field projection x.1, these projections can be pushed to the relational database so that unused fields are not retrieved and transferred.
Example 6.7.4: The rule for migrating projections to a relational database is implemented by MigrateProj in this example. The rule requires a function FullyProjected x N that traverses an expression N to determine whether x is always used within N in the context of a field projection and to determine what fields are being projected; it returns NONE if x is not always used in such a context; otherwise, it returns SOME L, where the list L contains all the fields being projected. This function is implemented in a simple way using the ContextSensitive rule generator from Example 6.7.3.


The MigrateProj rule is defined below. The function SQL. PushProj is one of the many support routines available in the current release of Kleisli that handle manipulation of SQL queries and other SYN abstract syntax objects.

Besides the four migration rules mentioned previously, Kleisli has various other rules, including reordering joins on two relational databases, parallelizing queries, and large-scale code motion, the description of which is omitted in the chapter due to space constraints.
6.8 USER INTERFACES
Kleisli is equipped with application programming interfaces for use with Java and Perl. It also has a graphical interface for non-programmers. These interfaces are described in this section.
6.8.1 Programming Language Interface
The high-level query language, sSQL, of the Kleisli system was designed to express traditional (nested relational) database-style queries. Not every query in bioinformatics falls into this class. For these non-database-style queries, some other programming languages can be a more convenient or more efficient means of implementation. The Pizzkell suite [19] of interfaces to the Kleisli exchange format was developed for various popular programming languages. Each of these interfaces in the Pizzkell suite is a library package for parsing data in Kleisli’s exchange format into an internal object of the corresponding programming language. It also serves as a means for embedding the Kleisli system into that programming language so that the full power of Kleisli is made available within that programming language. The Pizzkell suite currently includes CPL2Perl and CPL2Java, for Perl and Java.
In contrast to sSQL in Kleisli, which is a high-level interface that comes with a sophisticated optimizer and other database-style features, CPL2Perl has a different purpose and is at a lower level. Whereas sSQL is aimed at extraction, integration, and preparation of data for analysis, CPL2Perl is intended to be used for implementing analysis and textual formatting of the prepared data in Perl. Thus, CPL2Perl is a Perl module for parsing data conforming to the data exchange format of Kleisli into native Perl objects.
The main functions in CPL2Perl are divided into three packages:
1. The RECORD package simulates the record data type of the Kleisli exchange format by using a reference of Perl’s hash. Some functions are defined in this package:
![]() New is the constructor of a record. For example, to create a record such as(#anno_name: “db_xref”, #descr: “taxon: 10090”) in a Perl program, one writes:
New is the constructor of a record. For example, to create a record such as(#anno_name: “db_xref”, #descr: “taxon: 10090”) in a Perl program, one writes:
![]()
where $rec becomes the reference of this record in the Perl program.
![]() Project gets the value of a specified field in a record. For example,
Project gets the value of a specified field in a record. For example,
![]()
will return the value of the field #descr in the record referenced by $rec in the Perl program.
2. The LIST package simulates the list, set, and bag data type in the Kleisli data exchange format. These three bulk data types are to be converted as a reference of Perl’s list. Its main function is:
![]() new is the constructor of bulk data such as a list, a bag, or a set. It works the same way as a list initialization in Perl:
new is the constructor of bulk data such as a list, a bag, or a set. It works the same way as a list initialization in Perl:
![]()
where $1 will be the reference of this list in the Perl program.
3. The CPLIO package provides the interface to read data directly from a Kleisli-formatted data file or pipe. It supports both eager and lazy access methods. Some functions in this package are:
![]() Open1 opens the specified Kleisli-formatted data file and returns the handle of this file in Perl. It supports all the input-related features of the usual open operation of Perl, including the use of pipes. For example, the following expression opens a Kleisli-formatted file “sequences.val”:
Open1 opens the specified Kleisli-formatted data file and returns the handle of this file in Perl. It supports all the input-related features of the usual open operation of Perl, including the use of pipes. For example, the following expression opens a Kleisli-formatted file “sequences.val”:
![]()
![]() Openla is another version of the Openl function, which can take a string as input stream. The first parameter specifies the child process to execute and the second parameter is the input string. An example that calls Kleisli from CPL2Perl to extract accession numbers from a sequence file is expressed as follows:
Openla is another version of the Openl function, which can take a string as input stream. The first parameter specifies the child process to execute and the second parameter is the input string. An example that calls Kleisli from CPL2Perl to extract accession numbers from a sequence file is expressed as follows:

![]() Open2 differs from Openla in that it allows a program to communicate in both directions with Kleisli or other systems. It is parameterized by the Kleisli or other systems to call. It returns a list consisting of a reference of CPLIO object and an input stream that the requests can be sent into. For example:
Open2 differs from Openla in that it allows a program to communicate in both directions with Kleisli or other systems. It is parameterized by the Kleisli or other systems to call. It returns a list consisting of a reference of CPLIO object and an input stream that the requests can be sent into. For example:

![]() Parse is a function that reads all the data from an opened file until it can assemble a complete object or a semicolon (;) is found. The return value will be the reference of the parsed object in Perl. For example, printing the values of the field #accession of an opened file may be expressed by:
Parse is a function that reads all the data from an opened file until it can assemble a complete object or a semicolon (;) is found. The return value will be the reference of the parsed object in Perl. For example, printing the values of the field #accession of an opened file may be expressed by:

![]() LazyRead is a function that reads data lazily from an opened file. This function is used when the data type in the opened file is a set, a bag, or a list. This function only reads one element into memory at a time. Thus, if the opened file is a very big set, LazyRead is just the right function to access the records and print accession numbers:
LazyRead is a function that reads data lazily from an opened file. This function is used when the data type in the opened file is a set, a bag, or a list. This function only reads one element into memory at a time. Thus, if the opened file is a very big set, LazyRead is just the right function to access the records and print accession numbers:

The use of CPL2Perl for interfacing Kleisli to the Graphviz system is demonstrated in the context of the Protein Interaction Extraction System described in a paper by Wong [36]. Graphviz [37] is a system for automatic layout of directed graphs. It accepts a general directed graph specification, which is in essence a list of arcs of the form x → y, which specifies an arc is to be drawn from the node x to the node y.
Example 6.8.1: Assume that Kleisli produces a file $SPEC of type { (#actor: string, #interaction: string, #patient: string) } which describes a protein interaction pathway. The records express that an actor inhibits or activates a patient. The relevant parts of a Perl implementation of the module MkGif that accepts this file, converts it into a directed graph specification, invokes Graphviz to layout, and draws it as a GIF file $GIF using CPL2Perl is expressed as follows:

The first three lines establish the connections to the Kleisli file $SPEC and to the Graphviz program dot. The next few lines use the LazyRead and Project functions of CPL2Perl to extract each interaction record from the file and to format it for Graphviz to process. Upon finishing the layout computation, Graphviz draws the interaction pathway into the file $GIF.
This example, though short, demonstrates how CPL2Perl smoothly integrates the Kleisli exchange format into Perl. This greatly facilitates both the development of data drivers for Kleisli and the development of downstream processing (such as pretty printing) of results produced by Kleisli.
6.8.2 Graphical Interface
The Discovery Builder is a graphical interface to the Kleisli system designed for non-programmers by geneticXchange, Inc. This graphical interface facilitates the visualization of the source data as required to formulate the queries and generates the necessary sSQL codes. It allows users to see all available data sources and their associated meta-data and assists them in navigating and specifying their query on these sources with the following key functions:
![]() A graphical interface that can see all the relevant biological data sources, including meta-data—tables, columns, descriptions, etc.—and then construct a query as if the data were local
A graphical interface that can see all the relevant biological data sources, including meta-data—tables, columns, descriptions, etc.—and then construct a query as if the data were local
![]() Add new wrappers for any public or proprietary data sources, typically within hours, and then have them enjoined in any series of ad hoc queries that can be created
Add new wrappers for any public or proprietary data sources, typically within hours, and then have them enjoined in any series of ad hoc queries that can be created
![]() Execute the queries, which may join many data sources that can be scattered all over the globe, and get fresh result data quickly
Execute the queries, which may join many data sources that can be scattered all over the globe, and get fresh result data quickly
The Discovery Builder interface is presented in Figure 6.5.

6.9 OTHER DATA INTEGRATION TECHNOLOGIES
The brief description of several other approaches to bioinformatics data integration problems emphasizes Kleisli’s characteristics. The alternatives include Sequence Retrieval System (SRS) [38], DiscoveryLink [23], and OPM [22].
6.9.1 SRS
SRS [38] (also presented in Chapter 5) is marketed by LION Bioscience and is arguably the most widely used database query and navigation system for the life science community. It provides easy-to-use graphical user interface access to a broad range of scientific databases, including biological sequences, metabolic pathways, and literature abstracts. SRS provides some functionalities to search across public, in-house and in-licensed databases. To add a new data source into SRS, the data source is generally required to be available as a flat file, and a description of the schema or structure of the data source must be available as an Icarus script, which is the special built-in wrapper programming language of SRS. The notable exception to this flat file requirement on the data source is when the data source is a relational database. SRS then indexes this data source on various fields parsed and described by the Icarus script. A biologist then accesses the data by supplying some keywords and constraints on them in the SRS query language, and all records matching those keywords and constraints are returned. The SRS query language is primarily a navigational language. This query language has limited data joining capabilities based on indexed fields and has limited data restructuring capabilities. The results are returned as a simple aggregation of records that match the search constraints. In short, in terms of querying power, SRS is essentially an information retrieval system. It brings back records matching specified keywords and constraints. These records can contain embedded links a user can follow individually to obtain deeper information. However, it does not offer much help in organizing or transforming the retrieved results in a way that might be needed for setting up an analytical pipeline. There is also a Web browser-based interface for formulating SRS queries and viewing results. In fact, this interface of SRS is often used by biologists as a unified front end to access multiple data sources independently, rather than learning the idiosyncrasies of the original search interfaces of these data sources. For this reason, SRS is sometimes considered to serve “more of a user interface integration role rather than as a true data integration tool”[39].
In summary, SRS has two main strengths. First, because of the simplicity of flat file indexing, adding new data sources into the system with the Icarus scripting language is easy. In fact, several hundred data sources have been incorporated into SRS to date. Second, it has a nice user interface that greatly simplifies query formulation, making the system usable by a biologist without the assistance of a programmer. In addition, SRS has an extension known as Prisma designed for automating the process of maintaining an SRS warehouse. Prisma integrates the tasks of monitoring remote data sources for new data sets and downloading and indexing such data sets. On the other hand, SRS also has some weaknesses. First, it is basically a retrieval system that returns entries in a simple aggregation. To perform further operations or transformations on the results, a biologist has to do that by hand or write a separate post-processing program using some external scripting language like C or Perl, which is cumbersome. Second, its principally flat-file based indexing mechanism rules out the use of certain remote data sources—in particular, those that are not relational databases—and does not provide for straightforward integration with dynamic analysis tools. However, this latter shortcoming is mitigated by the Scout suite of applications marketed by LION Bioscience that are specifically designed to interact with SRS.
6.9.2 DiscoveryLink
DiscoveryLink [23] (also presented in Chapter 11) is an IBM product and, in principle, it goes one step beyond SRS as a general data integration system for biomedical data. The first thing that stands out—when DiscoveryLink is compared to SRS and more specialized integration solutions like EnsEMBL and GenoMax—is the presence of an explicit data model. This data model dictates the way DiscoveryLink users view the underlying data, the way they view results, as well as the way they query the data. The data model is the relational data model [17]. The relational data model is the de facto data model of most commercial database management systems, including the IBM’s DB2 database management system, upon which DiscoveryLink is based. As a result, DiscoveryLink comes with a high-level query language, SQL, that is a standard feature of all relational database management systems. This gives DiscoveryLink several advantages over SRS. First, not only can users easily express SQL queries that go across multiple data sources, which SRS users are able to do, but they can also perform further manipulations on the results, which SRS users are unable to do. Second, not only are the SQL queries more powerful and expressive than those of SRS, the SQL queries are also automatically optimized by DB2. Query optimization allows users to concentrate on getting their queries right without worrying about getting them fast.
However, DiscoveryLink still has to overcome difficulties. The first reason is that DiscoveryLink is tied to the relational data model. This implies that every piece of data it handles must be a table of atomic objects, such as strings and numbers. Unfortunately, most of the data sources in biology are not that simple and are deeply nested. Therefore, there is some impedance mismatch between these sources and DiscoveryLink. Consequently, it is not straightforward to add new data sources or analysis tools into the system. For example, to put the Swiss-Prot [40] database into a relational database in the third normal form would require breaking every Swiss-Prot record into several pieces in a normalization process. Such a normalization process requires a certain amount of skill. Similarly, querying the normalized data in DiscoveryLink requires some mental and performance overhead, as the user needs to figure out which part of Swiss-Prot has gone to which of the pieces and to join some of the pieces back again to reconstruct the entry. The second reason is that DiscoveryLink supports only wrappers written in C++, which is not the most suitable programming language for writing wrappers. In short, it is not straightforward to extend DiscoveryLink with new sources. In addition, DiscoveryLink does not store nested objects in a natural way and is very limited in its capability for handling long documents. It also has limitations as a tool for creating and managing data warehouses for biology.
6.9.3 Object-Protocol Model (OPM)
Developed at Lawrence-Berkeley National Labs, OPM [22] is a general data integration system. OPM was marketed by GeneLogic, but its sales were discontinued some time ago. It goes one step beyond DiscoveryLink in the sense that it has a more powerful data model, which is an enriched form of the entity-relationship data model [41]. This data model can deal with the deeply nested structure of biomedical data in a natural way. Thus, it removes the impedance mismatch. This data model is also supported by an SQL-like query language that allows data to be seen in terms of entities and relationships. Queries across multiple data sources, as well as transformation of results, can be easily and naturally expressed in this query language. Queries are also optimized. Furthermore, OPM comes with a number of data management tools that are useful for designing an integrated data warehouse on top of OPM.
However, OPM has several weaknesses. First, OPM requires the use of a global integrated schema. It requires significant skill and effort to design a global integrated schema well. If a new data source needs to be added, the effort needed to re-design the global integrated schema potentially goes up quadratically with respect to the number of data sources already integrated. If an underlying source evolves, the global integrated schema tends to be affected and significant re-design effort may be needed. Therefore, it may be costly to extend OPM with new sources. Second, OPM stores entities and relationships internally using a relational database management system. It achieves this by automatically converting the entities and relationships into a set of relational tables in the third normal form. This conversion process breaks down entities into many pieces when stored. This process is transparent to OPM users, so they can continue to think and query in terms of entities and relationships. Nevertheless, the underlying fragmentation often causes performance problems, as many queries that do not involve joins at the conceptual level of entities and relations are mapped to queries that evoke many joins on the physical pieces to reconstruct broken entities. Third, OPM does not have a simple format to exchange data with external systems. At one stage, it interfaces to external sources using the Common Object Request Broker Architecture (CORBA). The effort required for developing CORBA-compliant wrappers is generally significant [42]. Furthermore, CORBA is not designed for data-intensive applications.
6.10 CONCLUSIONS
In the era of genome-enabled, large-scale biology, high-throughput technologies from DNA sequencing, microarray gene expression and mass spectroscopy, to combinatory chemistry and high-throughput screening have generated an unprecedented volume and diversity of data. These data are deposited in disparate, specialized, geographically dispersed databases that are heterogeneous in data formats and semantic representations. In parallel, there is a rapid proliferation of computational tools and scientific algorithms for data analysis and knowledge extraction. The challenge to life science today is how to process and integrate this massive amount of data and information for research and discovery. The heterogeneous and dynamic nature of biomedical data sources presents a continuing challenge to accessing, retrieving, and integrating information across multiple sources.
Many features of the Kleisli system [2, 5, 43] are particularly suitable for automating the data integration process. Kleisli employs a distributed and federated approach to access external data sources via the wrapper layer, and thus can access the most up-to-date data on demand. Kleisli provides a complex nested internal data model that encompasses most of the current popular data models including flat files, HTML, XML, and relational databases, and thus serves as a natural data exchanger for different data formats. Kleisli offers a robust query optimizer and a powerful and expressive query language to manipulate and transform data, and thus facilitates data integration. Finally, Kleisli has the capability of converting relational database management systems such as Sybase, MySQL, Oracle, DB2, and Informix into nested relational stores, thus enabling the creation of robust warehouses of complex biomedical data. Leveraging the capabilities of Kleisli leads to the development of the query scripts that give us a high-level abstraction beyond low-level codes to access a combination of the relevant data and the right tools to solve the right problem.
Kleisli embodies many of the advances in database query languages and in functional programming. The first is its use of a complex object data model in which sets, bags, lists, records, and variants can be flexibly combined. The second is its use of a high-level query language that allows these objects to be easily manipulated. The third is its use of a self-describing data exchange format, which serves as a simple conduit to external data sources. The fourth is its query optimizer, which is capable of many powerful optimizations. It has had significant impact on data integration in bioinformatics. Indeed, since the early Kleisli prototype was applied to bioinformatics, it has been used efficiently to solve many bioinformatics data integration problems.
REFERENCES
[1] revision 2.0 National Center for Biotechnology Information (NCBI). NCBI ASN.l Specification. Bethesda, MD: National Library of Medicine; 1992.
[2] Wong, L. Kleisli: A Functional Query System. Journal of Functional Programming. 2000;10(no. 1):19–56.
[3] Backus, J. Can Programming Be Liberated from Von Neumann Style? A Functional Style and Its Algebra of Programs. Communications of the ACM. 1978;21(no. 8):613–641.
[4] Darlington, J. An Experimental Program Transformation and Synthesis System. Artificial Intelligence. 1981;16(no. 1):1–46.
[5] Davidson, S., Overton, C., Tannen, V., et al. BioKleisli: A Digital Library for Biomedical Researchers. International Journal of Digital Libraries. 1997;1(no. 1):36–53.
[6] Robbins R.J., ed. Report of the Invitational DOE Workshop on Genome Informatics. Baltimore, MD. 1993:26–27.
[7] supplement Pearson, P., Matheson, N.W., Flescher, D.L., et al. The GDB Human Genome Data Base Anno 1992. Nucleic Acids Research. 1992;20:2201–2206.
[8] supplement Burks, C., Cinkosky, M.J., Fischer, W.M. GenBank. Nucleic Acids Research. 1992;20:2065–2069.
[9] Schuler, G.D., Epstein, J.A., Ohkawa, H., et al. Entrez: Molecular Biology Database and Retrieval System. Methods in Enzymology. 1996;266:141–162.
[10] Bailey, L.C., Jr., Fischer, S., Schug, J., et al. GALA: Framework Annotation of Genomic Sequence. Genome Research. 1998;8(no. 3):234–250.
[11] Baker, P.G., Brass, A., Bechhofer, S. TAMBIS: Transparent Access to Multiple Bioinformatics Information Sources. Intelligent Systems for Molecular Biology. 1998;6:25–34.
[12] Goble, C.A., Stevens, R., Ng, G. Transparent Access to Multiple Bioinformatics Information Sources. IBM Systems Journal. 2001;40(no. 2):532–552.
[13] Schoenbach, C., Koh, J., Sheng, X. FIMM: A Database of Functional Molecular Immunology. Nucleic Acids Research. 2000;28(no. 1):222–224.
[14] Buneman, P., Libkin, L., Suciu, D. Comprehension Syntax. SIGMOD Record. 1994;23(no. 1):87–96.
[15] Wadler, P. Comprehending Monads. Mathematical Structures in Computer Science. 1992;2(no. 4):461–493.
[16] Buneman, P., Naqui, S., Tannen, V., et al. Principles of Programming With Complex Objects and Collection Types. Theoretical Computer Science. 1995;149(no. 1):3–48.
[17] Codd, E.F. A Relational Model for Large Shared Data Bank. Communications of the ACM. 1970;13(no. 6):377–387.
[18] Walsh, S., Anderson, M., Cartinhour, S.W., et al. ACEDB: A Database for Genome Information. Methods of Biochemical Analysis. 1998;39:299–318.
[19] Wong, L. Kleisli: Its Exchange Format, Supporting Tools, and an Application in Protein Interaction Extraction. In: Proceedings of the First IEEE International Symposium on Bio-Informatics and Biomedical Engineering. Los Alamitos, CA: IEEE Computer Society; 2000:21–28.
[20] International Standards Organization (ISO). Standard 8824: Information Processing Systems. Open Systems Interconnection. Specification of Abstraction Syntax Notation One (ASN.l). Geneva, Switzerland: ISO; 1987.
[21] Hubbard, T., Barker, D., Birney, E. The ENSEMBL Genome Database Project. Nucleic Acids Research. 2002;30(no. 1):38–41.
[22] Chen, I.M.A., Markowitz, V.M. An Overview of the Object-Protocol Model (OPM) and OPM Data Management Tools. Information Systems. 1995;20(no. 5):393–418.
[23] Haas, L.M., Schwarz, P.M., Kodali, P. Disco very Link: A System for Integrated Access to Life Sciences Data Sources. IBM Systems Journal. 2001;40(no. 2):489–511.
[24] Tannen, V., Buneman, P., Nagri, S. Structural Recursion as a Query Language. In: Proceedings of the Third International Workshop on Database Programming Languages. San Francisco: Morgan Kaufmann; 1991:9–19.
[25] Tannen, V., Subrahmanyam, R., Logical and Computational Aspects of Programming with Sets/Bags/Lists, Proceedings of the 18th International Colloquium on Automata, Languages, and Programming, Lecture Note in Computer Science. Berlin, Germany: Springer-Verlag; 1991;vol. 510:60–75.
[26] Suciu, D. Bounded Fixpoints for Complex Objects. Theoretical Computer Science. 1997;176(no.1–2):283–328.
[27] Dong, G., Libkin, L., Wong, L. Local Properties of Query Languages. Theoretical Computer Science. 2000;239(no. 2):277–308.
[28] Libkin, L., Wong, L. Query Languages for Bags and Aggregate Functions. Journal of Computer and System Sciences. 1997;55(no. 2):241–272.
[29] Suciu, D., Wong, L., On Two Forms of Structural Recursion, Proceedings of the Fifth International Conference on Database Theory, Lecture Notes in Computer Science. Berlin, Germany: Springer-Verlag; 1995;vol. 83:111–124.
[30] Altschul, S.F., Gish, W. Local Alignment Statistics. Methods in Enzymology. 1996;266:460–480.
[31] Altschul, S.F., Madden, T.L., Schaffer, A.A., Blast, Gapped. PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research. 1997;25(no. 17):3389–3402.
[32] Thompson, J.D., Higgins, D.G., Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Positions-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Research. 1994;22:4673–4680.
[33] Falquet, L., Pagni, M., Bucher, P., et al, The PROSITE Database, Its Status in 2002. Nucleic Acids Research. 2002;30(no. 1):235–238. sib.ch/cgibin/PFSCAN
[34] Murzin, A., Brenner, S.E., Hubbard, T., et al. SCOP: A Structural Classification of Protein Database for the Investigation of Sequences and Structures. Journal of Molecular Biology. 1995;247(no. 4):536–540.
[35] Goldberg, A., Paige, R. Stream Processing. In: Proceedings of the ACMSymposium on LISP and Functional Programming. New York: ACM; 1984:53–62.
[36] Wong, L. PIES, A Protein Interaction Extraction System. In: Proceedings of the Pacific Symposium in Biocomputing. Singapore: World Scientific; 2001:520–531.
[37] Gansner, E.R., Koutsofios, E., North, S.C., et al. A Technique for Drawing Directed Graphs. IEEE Transactions on Software Engineering. 1993;19(no. 3):214–230.
[38] Etzold, T., Argos, P. SRS: Information Retrieval System for Molecular Biology Data Banks. Methods in Enzymology. 1996;266:114–128.
[39] 3rd Millennium Inc. Practical Data Integration in Biopharmaceutical R & D: Strategies and Technologies. Cambridge, MA: 3rd Millennium; 2002.
[40] Bairoch, A., Apweiler, R. The SWISS-PROT Protein Sequence Data Bank and Its Supplement TrEMBL in 1999. Nucleic Acids Research. 1999;27(no. 1):49–54.
[41] Chen, P.P.S. The Entity-Relationship Model: Toward a Unified View of Data. ACM Transactions on Database Systems. 1976;1(no. 1):9–36.
[42] Selletin, J., Mitschang, B. Data-Intensive Intra- & Internet Applications: Experiences Using Java and CORBA in the World Wide Web. In: Proceedings of the Fourteenth IEEE International Conference on Data Engineering. Los Alamitos, CA: IEEE Computer Science; 1998:302–311.
[43] Wong, L. The Functional Guts of the Kleisli Query System. In: Proceedings of the Fifth ACM SIGPLAN International Conference on Functional Programming. New York: ACM; 2000:1–10.
1Earlier versions of the Kleisli system supported only a query language based on comprehension syntax called Collection Programming Language (CPL) [2]. Now, both CPL and sSQL are available.
2Functional programming languages are programming languages that emphasize a particular paradigm of programming technique known as functional programming [3, 4]. In this paradigm, all programs are expressed as mathematical functions and are generally free from side effects. Examples of functional programming languages are LISP, Haskell, and SML. Some fundamental ideas in functional programming languages, such as garbage collection, have also been borrowed by other modern programming languages such as Java.
3Bulk data types refer to data types that are collections of objects. Examples of bulk data types are sets, bags, lists, and arrays.
4Information about the GAIA project is available at http://www.cbil.upenn.edu/gaia2/gaia and [10].
5Information about Discovery Hub is available at http://www.geneticxchange.com.
6s21 denotes a function that converts a set into a list. list-sum is a function to sum a list of numbers. The function ml-get-count-general accesses MEDLINE and computes the number of MEDLINE reports matching a given keyword, whereas ml-get-abstract-by-uid is a function that accesses MEDLINE to retrieve reports given a unique identifier, and webomim-get-id accesses the OMIM database to obtain unique identifiers of OMIM reports matching a keyword. webomim-get-detail is a function that accesses OMIM to retrieve reports given a unique identifier. hugo-get-by-symbol is a function that accesses the HUGO database and returns HUGO reports matching a given gene name.