
Issues to Address While Designing a Biological Information System
Life science has experienced a fundamental revolution from traditional in vivo discovery methods (understanding genes, metabolic pathways, and cellular mechanisms) to electronic scientific discovery consisting in collecting measurement data through a variety of technologies and annotating and exploring the resulting electronic data sets. To cope with this dramatic revolution, life scientists need tools that enable them to access, integrate, mine, analyze, interpret, simulate, and visualize the wealth of complex and diverse electronic biological data. The development of adequate technology faces a variety of challenges. First, there exist thousands of biomedical data sources: There are 323 relevant public resources in molecular biology alone [1]. The number of biological resources increases at great pace. Previous lists of key public resources in molecular biology contained 203 data sources in 1999 [2], 226 in 2000 [3], and 277 in 2001 [4]. Access to these data repositories is fundamental to scientific discovery. The second challenge comes from the multiple software tools and interfaces that support electronic-based scientific discovery. An early report from the 1999 U.S. Department of Energy Genome Program meeting [5] held in Oakland, California identified these challenges with the following statement:
Genome-sequencing projects are producing data at a rate exceeding current analytical and data-management capabilities. Additionally, some current computing problems are expected to scale up exponentially as the data increase. [5]
The situation has worsened since, whereas the need for technology to support scientific discovery and bioengineering has significantly increased. Chapter 1 covers the reasons why, ultimately, all of these resources must be combined to form a comprehensive picture. Chapter 2 claims that this challenge may well constitute the backbone of 21st century life science research. In the past, the specific research and development of geographical and spatial data management systems led to the emergence of an important and very active geographic information systems (GIS) community. Likewise, the field of biological information systems (BIS) aiming to support life scientists is now emerging.
To develop biological information systems, computer scientists must address the specific needs of life scientists. The identification of the specifications of computer-aided biology is often impeded by difficulty of communication between life scientists and computer scientists. Two main reasons can be identified. First, life and computer scientists have radically different perspectives in their development activities. These discrepancies can be explained by comparing the design process in engineering and in experimental sciences. A second reason for misunderstanding results from their orthogonal objectives. A computer scientist aims to build a system, whereas a life scientists aims to corroborate an hypothesis. These viewpoints are illustrated in the following.
Engineering vs. Experimental Science
Software development has an approach similar to engineering. First, the specifications (or requirements) of the system to be developed are identified. Then, the development relies on a long initial design phase when most of the cases, if not all, are identified and offered a solution. Only then is a prototype implemented. Later, iterations of the loop design ↔ implementation aim to correct the implementation’s failure to perform effectively the requirements and to extend, significantly, the implementation to new requirements. These iterations are typically expressed through codified versioning of the prototype. In practice, initial design phases are typically shortened because of drastic budget cuts and a hurry to market the product. However, short design phases often cause costly revisions that could have been avoided with appropriate design effort.
Bioinformatics aims to support life scientists in the discovery of new biological insights as well as to create a global perspective from which unifying principles in biology can be discerned. Scientific discovery is experimental and follows a progress track blazed by experiments designed to corroborate or fail hypotheses. Each experiment provides the theory with additional material and knowledge that builds the entire picture. An hypothesis can be seen as a design step, whereas an experiment is an implementation of the hypothesis. Learning thus results from multiple iterations of design ↔ implementation, where each refinement of an hypothesis is motivated by the failure of the previous implementation.
These two approaches seem very similar, but they vary by the number of iterations of design ↔ implementation. When computer scientists are in the design phase of developing a new system for life scientists, they often have difficulties in collecting use cases and identifying the specifications of the system prior to implementation. Indeed, life scientists are likely able to provide just enough information to build a prototype, which they expect to evaluate to express more requirements for a better prototype, and so on. This attitude led a company proposing to build a biological data management system to offer to “build a little, test a little” to ensure meeting the system requirements. Somehow the prototype corresponds to an experiment for a life scientist. Understanding these two dramatically different approaches to design is mandatory to develop useful technology to support life scientists.
Generic System vs. Query-Driven Approach
Computer scientists aim to build systems. A system is the implementation of an approach that is generic to many applications having similar characteristics. When provided with use cases or requirements for a new system, computer scientists typically abstract them as much as possible to identify the intrinsic characteristics and therefore design the most generic approach that will perform the requirements in various similar applications.
Life scientists, in their discovery process, are motivated by an hypothesis they wish to validate. The validation process typically involves some data sets extracted from identified data sources and follows a pre-defined manipulation of the collected data. In a nutshell, a validation approach corresponds to a complex query asked against multiple and often heterogeneous data sources. Life scientists have a query-driven approach.
These two approaches are orthogonal but not contradictory. Computer scientists present the value of their approach by illustrating the various queries the system will answer, whereas life scientists value their approach by the quality of the data set obtained and the final validation of the hypothesis. This orthogonality also explains the legacy in bioinformatics implementations, which mostly consist of hard-coded queries that do not offer the flexibility of a system as explained and illustrated in Chapters 1 and 3. This legacy problem is addressed in Section 4.1.2.
This chapter does not aim to present or compare the systems that will be described in the later chapters of this book. Instead it is devoted to issues specific to data management that need to be addressed when designing systems to support life science. As with any technology, data management assumes an ideal world upon which most systems are designed. They appear to suit the needs of large corporate usage such as banking; however, traditional technology fails to adjust properly to many new technological challenges such as Web data management and scientific data management. The following sections introduce these issues.
Section 4.1 presents some of the characteristics of available scientific data and technology. Section 4.2 is devoted to the first issue that traditional data management technology needs to address: changes. Section 4.3 addresses issues related to biological queries, whereas Section 4.4 focuses on query processing. Finally, data visualization is addressed in Section 4.5. Bioinformaticians should find a variety of illustrations of the reasons why BIS need innovative solutions.
4.1 LEGACY
Scientific data has been collected in electronic form for many years. While new data management approaches are designed to provide the basis for future homogeneous collection, integration, and analysis of scientific data, they also need to integrate existing large data repositories and a variety of applications developed to analyze them. Legacy data and tools may raise various difficulties for their integration that may affect the design of BIS.
4.1.1 Biological Data
Scientific data are disseminated in myriad different data sources across disparate laboratories, available in a wide variety of formats, annotated, and stored in flat files and relational or object-oriented databases. Access to heterogeneous biological data sources is mandatory to scientists. A single query may involve flat files (stored locally or remotely) such as GenBank [6] or Swiss-Prot [7], Web resources such as the Saccharomyces Genome Database [8], GeneCards [9], or the references data source PubMed [10]. A list of useful biological data sources is given in the Appendix. These sources are mostly textual and of restricted access facilities. Their structure varies from ASN. 1 data exchange format to poorly structured hypertext markup language (HTML) and extensible markup language (XML) formats. This variety of repositories justifies the need to evoke data sources rather than databases. This chapter only refers to a database when the underlying system is a database management system (a relational database system, for example). A system based on flat files is not a database.
Unlike data hosted in a database system, scientific data is maintained by life scientists through user-friendly interfaces that offer great flexibility to add and revise data in these data sources. However, this flexibility often affects data quality dramatically. First, data sources maintained by a large community such as GenBank contain large quantities of data that need to be curated. This explains the numerous overlapping data sources sometimes found aiming to complete or correct data from existing sources. As importantly, data organization also suffers from this wide and flexible access: Data fields are often completed with different goals and objectives, fields are missing, and so on. This flexibility is typically provided when the underlying data representation is a formatted file with no types or constraints checking. Unfortunately this variety of data representation makes it difficult to use traditional approaches as explained in Section 4.2.3.
The quantity of data sources to be exploited by life scientists is overwhelming. Each year the number of publicly available data sources increases significantly: It rose 43% between 1999 and 2002 for the key molecular data sources [1, 2]. In addition to this proliferation of sources, the quantity of data contained in each data source is significantly large and also increasing. For example, as of January 1, 2001, GenBank [6] contained 11,101,066,288 bases in 10,106,023 sequences, and its growth continues to be exponential, doubling every 14 months [11]. While the number of distinct human genes appears to be smaller than expected, in the range of 30,000–40,000 [12, 13], the distinct human proteins in the proteome are expected to number in the millions due to the apparent frequency of alternative splicing, ribonucleic acid (RNA) editing, and post-translational modification [14, 15]. As of May 4, 2001, Swiss-Prot contained 95,674 entries, whereas PubMed contained more than 11 million citations. Managing these large data sets efficiently will be critical in the future. Issues of efficient query processing are addressed in Section 4.4.
Future collaborations between computer and life scientists may improve the collection and storage of data to facilitate the exploitation of new scientific data. However, it is likely that scientific data management technology will need to address issues related to the characteristics of the large existing data sets that constitute part of the legacy of bioinformatics.
4.1.2 Biological Tools and Workflows
Scientific resources include a variety of tools that assist life scientists in searching, mining, and analyzing the proliferation of data. Biological tools include basic biosequence analyses such as FASTA, BLAST, Clustal, Mfold, Phylip, PAUP, CAP, and MEGA. A data management system not integrating these useful tools would offer little support to life scientists. Most of these tools can be used freely by loading their code onto a computer from a Web site. A list of 160 free applications supporting biomolecular biology is provided in Misener and Krawetz’s Bioinformatics: Methods and Protocols [16]. The first problem is the various platforms used by life scientists: In 1998, 30-50% of biologists used Macintosh computers, 40-70% used a PC running any version of Microsoft Windows, and less than 10% used UNIX or LINUX [16]. Out of the 160 applications listed in Misener and Krawetz’s book, 107 run on a PC, 88 on a Macintosh, and 42 on other systems such as UNIX. Although some applications (27 listed in the previously mentioned book) are made available for all computer systems, most of them only run on a single system. The need for integration of applications, despite the system for which they may be designed, motivated the idea of the grid as explained in Section 4.3.4.
Legacy tools also include a variety of hard-coded scripts in languages such as Perl or Python that implement specific queries, link data repositories, and perform a pre-defined sequence of data manipulation. Scripting languages were used extensively to build early bioinformatics tools. However, they do not offer expected flexibility for re-use and integration with other functions. Most legacy integration approaches were developed using workflows. Workflows are used in business applications to assess, analyze, model, define, and implement the core business processes of an organization (or other entity). A workflow approach automates the business procedures where documents, information, or tasks are passed between participants according to a defined set of rules to achieve, or contribute to, an overall business goal. In the context of scientific applications, a workflow approach may address overall collaborative issues among scientists, as well as the physical integration of scientific data and tools. The procedural support a workflow approach provides follows the query-driven design of scientific problems presented in the Introduction. In such an approach, the data integration problem follows step-by-step the single user’s query execution, including all necessary “business rules” such as security and semantics. A presentation of workflows and their model is provided online by the Workflow Management Coalition [17].
The integration of these tools and query pipelines into a BIS poses problems that are beyond traditional database management as explained in Section 4.3.4.
4.2 A DOMAIN IN CONSTANT EVOLUTION
A BIS must be designed to handle a constantly changing domain while managing legacy data and technology. Traditional data management approaches are not suitable to address constant changes (see Section 4.2.1). Two problems are critical to address for scientific data management: changes in data representation (see Section 4.2.2) and data identification (see Section 4.2.4). The approach presented in Chapter 10 addresses specifically these problems with gene expression data.
4.2.1 Traditional Database Management and Changes
The main assumption of traditional data management approaches relies on a predefined, unchangeable system of data organization. Traditional database management systems are of three kinds: relational, object-relational, or object-oriented. Relational database systems represent data in relations (tables). Object-relational systems provide a more user-friendly data representation through classes, but they rely on an underlying relational representation. Object-oriented databases organize data through classes. For the sake of simplicity, only relational databases are considered in this section because most of the databases currently used by life scientists are relational, and similar remarks could be made for all traditional systems.
Data organization includes the relations and attributes that constitute a relational database schema. When the database schema is defined, it can be populated by data to create an instance of the schema, in other words, a database. Each row of a relation is called a tuple. When a database has been defined, transactions may be performed to update the data contained in the database. They consist of insertions (adding new tuples in relations), deletions (removing tuples from relations), and updates (transforming one or more components of tuples in relations). All traditional database systems are designed to support transactions on their data; however, they support few changes in the data organization. Changes in the data organization include renaming relations or attributes, removing or adding relations or attributes, merging or splitting relations or attributes, and so on. Some transformations such as renaming are rather simple, and others are complex. Traditional database systems are not designed to support complex schema transactions. Typically, a change in the data organization of a database is performed by defining a new schema and loading the data from the database to an instance of the new schema, thus creating a new database. Clearly this process is tedious and not acceptable when such changes have to be addressed often. Another approach to the problem of restructuring is to use a view mechanism that offers a new schema to users as a virtual schema when the underlying database and schema have not changed. All user interfaces provide access to the data as they are defined in the view and no longer as they are defined in the database. This approach offers several advantages, including the possibility of providing customized views of databases. A view may be limited to part of the data for security reasons, for example. However, this approach is rather limited as the transactions available through the view may be restricted.
Another aspect of traditional database systems relies on a pre-defined identity. Objects stored in a relational database can be identified by a set of attributes that, together, characterize the object. For example, the three attributes—first name, middle name, and last name—can characterize a person. The set of characterizing attributes is called a primary key. The concept of primary key relies on a characterization of identity that will not change over time. No traditional database system is designed to address changes in identification, such as tracking objects that may have changed identity over time.
4.2.2 Data Fusion
Data fusion corresponds to the need to integrate information acquired from multiple sources (sensors, databases, information gathered by humans, etc.). The term was first used by the military to qualify events, activities, and movements to be correlated and analyzed as they occurred in time and space to determine the location, identity, and status of individual objects (equipment and units), assess the situation, qualitatively or quantitatively determine threats, and detect patterns in activity that would reveal intent or capability.
Scientific data may be collected through a variety of instruments and robots performing microarrays, mass spectrometry, flow cytometry, and other procedures. Each instrument needs to be calibrated, and the calibration parameters may affect the data significantly. Different instruments may be used to perform similar tasks and collect data to be integrated in a single data set for analysis. The analysis is performed over time upon data sets disparate by the context of their collection. The analysis must be tempered by parameters that directly affect the quality of the data. A similar problem is presented in Section 10.4.3 in the context of probe arrays and gene expression. A traditional database approach requires the complete collection of measurement data and all parameters to allow the expression of the complex queries that enable the analysis of the disparate data set. Should any information be missing (NULL in a table), the system ignores the corresponding data, an unacceptable situation for a life scientist. In addition, the use of any new instrument that requires the definition of new parameters may affect the data organization, as well as make the fusion process more complex. The situation is made even more complex by the constant evolution of the protocols. Their new specifications often change the overall data organization: Attributes may be added, split, merged, removed, or renamed. Traditional database systems’ difficulty with these issues of data fusion explains the current use of Microsoft Excel spreadsheets and manual computation to perform the integration prior to analysis. The database system is typically used as a storage device.
Can a traditional database system be adjusted to handle these constant and complex changes in the data organization? It is unlikely. Indeed, all traditional approaches rely strongly on a pre-defined and stale data organization. A BIS shall offer great flexibility in the data organization to meet the needs of life scientists. New approaches must be designed to enable scientific data fusion. A solution is to relax the constraint on the data representation, as presented in the following section.
4.2.3 Fully Structured vs. Semi-Structured
Traditional database approaches are too structured: When the schema is defined, it is difficult to change it, and they do not support the integration of similar, but disparate, data sets. A solution to this need for adherence to a structure is offered by the semi-structured approach. In the semi-structured approach, the data organization allows changes such as new attributes and missing attributes. Semi-structured data is usually represented as an edge-labeled, rooted, directed graph [18–20]. Therefore, a system handling semi-structured data does not assume a given, pre-defined data representation: A new attribute name is a new labeled edge, a new attribute value is a new edge in the graph, and so on. Such a system should offer greater flexibility than traditional database systems. An example of representation of semi-structured data is XML, the upcoming standard for data exchange on the Web designed by the World Wide Web Consortium (W3C). XML extends the basic tree-based data representation of the semi-structured model by ordering elements and providing various levels of representation such as XML Schema [21–23]. These additional characteristics make XML data representation significantly less flexible than the original semi-structured data model. Fully structured data representation, semi-structured data representation, and XML are presented in Data on the Web [24].
There are currently two categories of XML management systems: XML-enabled and native XML. The first group includes traditional database systems extended to an XML interface for collection and publication. However, the underlying representation is typically with tables. Examples of XML-enabled systems are Oracle9i1 and SQL Server 2000.2 These systems were mostly designed to handle Business-to-Business (B2B) and Business-to-Customer (B2C) business tasks on the Web. They have not yet proven useful in scientific contexts. Native XML systems such as Tamino,3 ToX,4 and Galax5 rely on a real semi-structured approach and should provide a flexibility interesting in the context of scientific data management.
Because of XML’s promising characteristics, and because it is going to be the lingua franca for the Web, new development for BIS should take advantage of this new technology. A system such as KIND, presented in Chapter 12, already exploits XML format. However, the need for semantic data integration in addition to syntactic data integration (as illustrated in Section 4.2.5) limits the use of XML and its query language in favor of approaches such as description logics.
4.2.4 Scientific Object Identity
Scientific objects change identification over time. Data stored in data sources can typically be accessed with knowledge of their identification or other unique characterization initially entered into the data bank. Usually, each object of interest has a name or an identifier that characterizes it. However, a major problem arises when a given scientific object, such as a gene, may possess as many identifiers as there are data sources that contain information about it. The challenge is to manage these semantic heterogeneities at data access, as the following example illustrates.
Gene names change over time. For example, the Human Gene Nomenclature Database (HUGO) [25] contains 13,594 active gene symbols, 9635 literature aliases, and 2739 withdrawn symbols. In HUGO, SIR2L1 (withdrawn) is a synonym to SIRT1 (the current approved HUGO symbol) and sir2-like 1. P53 is a withdrawn HUGO symbol and an alias for TP53 (current approved HUGO symbol). When a HUGO name is removed, not all data sources containing the name are updated. Some information, such as the content of PubMed, will actually never be updated.
Table 4.1 illustrates the discrepancies found when querying biological data sources with equivalent (but withdrawn or approved) HUGO names in November 2001. The Genome DataBase (GDB) is the official central repository for genomic mapping data resulting from the Human Genome Initiative [26]. Gen Atlas [27, 28] provides information relevant to the mapping of genes, diseases, and markers. Online Mendelian Inheritance in Man (OMIM) [29, 30] is a catalog of human genes and genetic disorders. GeneCards [9] is a data source of human genes, their products, and their involvement in diseases. LocusLink [31, 32] provides curated sequence and descriptive information about genetic loci.
TABLE 4.1
Number of entries retrieved with HUGO names from GDB, GenAtlas, OMIM, GeneCards, and LocusLink.
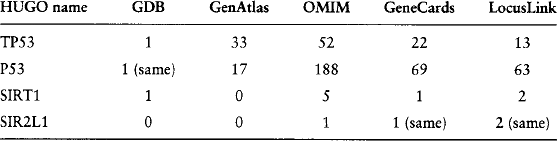
Querying GDB with TP53 or its alias P53 does not affect the result of the query. However, SIRT1 returns a single entry, whereas its alias does not return any entry. Gen Atlas returns more entries for the approved symbol TP53 than the withdrawn symbol P53. The question, then, is to determine if the entries corresponding to a withdrawn symbol are always contained in the set of entries returned for an approved symbol. This property does not hold in OMIM. OMIM returns many more entries for the withdrawn symbol P53 than the approved symbol TP53. However, it shows opposite behavior with SIRT1 and its alias. This demonstrates that even with the best understood and most commonly accepted characteristic of a gene—its identification—alternate identifier values need to be used when querying multiple data sources to get complete and consistent results. This problem requires significant domain expertise to resolve but is critical to the task of obtaining a successful, integrated biological information system.
The problem of gene identity is made more complex when the full name, alternative titles, and description of a gene are considered. Depending on the number of data sources that describe the gene, it may have that many equivalent source identifiers. For example, SIRT1 is equivalent to the full name (from HUGO) of sirtuin (silent mating type information regulation 2, S. cerevisiae, homolog) 1. This is also its description in LocusLink, but it has the following alternative title in OMIM:SIR2, S. CEREVISIAE, HOMOLOG-LIKE 1. These varying qualifications can often be easily discerned by humans, but they prove to be very difficult when automated. Here, too, extensive domain expertise is needed to determine that these descriptions each represent the very same gene, SIRT1. Although this is difficult, it does not describe the entire problem. There are as many identifiers to SIRT1 as there are data sources describing the gene. For example, SIRT1 also corresponds to 604479 (OMIM number), AF083106 (GenBank accession number), and 9956524 (GDB ID). Even UniGene clusters may have corresponding aliases. For example, Hs. 1846 (the UniGene cluster for P53) [33] is an alias for Hs .103997 (primary cluster for TP53).
Existing traditional approaches do not address the complex issues of scientific object identity. However, recent work on ontologies may provide solutions to these issues.
4.2.5 Concepts and Ontologies
An ontology is a collection of vocabulary words that define a community’s understanding of a domain. Terms are labels for concepts, which reside in a lattice of relationships between concepts. There have been significant contributions to the specification of standards and ontologies for the life science community as detailed in Chapter 2. In addition, some BIS were designed to provide users’ access to data as close as possible to their understanding. The system [34] based on the Object Protocol Model (OPM), developed at Lawrence Berkeley Laboratory and later extended and maintained at Gene Logic, provides data organization through classes and relationships to the user (see Chapter 10). The most successful such approach is TAMBIS (see Chapter 7), which was developed to allow users to access and query their data sets through an ontology. Such approaches are friendly because they allow life scientists to visualize the data sets through their understanding of the overall organization (concepts and relationships) as opposed to an arbitrary and often complex database representation with tables or a long list of tags of a flat file.
A solution to the problem of capturing equivalent representations of objects consists in using concepts. For example, a concept gene can have a primary identity (its approved HUGO symbol) and equivalent representations (withdrawn HUGO names, aliases, etc.). There are many ways to construct these equivalent classes. One way consists in collecting these multiple identities and materializing them within a new data source. This approach was partially completed in LENS, which was developed at the University of Pennsylvania, and GeneCards. This first approach captures the expertise of life scientists, and these data sources are usually well curated. To make this task scalable to all the scientific objects of interest, specific tools need to be developed to enhance and assist scientists in the task of managing the identity of scientific objects. While this expertise could be materialized within a new data source, it is critical that it is used by a BIS to alert the biologist when it recognizes alternate identifiers that could lead to incomplete or inconsistent results. This approach could use entity matching tools [35, 36] that capture similarities in retrieved objects and are appropriate for matching many of the functional attributes such as description or alternate titles.
Recent work in the context of the Semantic Web activity of the W3C may develop more advanced technology to provide users a sound ontology layer to integrate their underlying biological resources. However, these approaches do not yet provide a solution to the problem of capturing equivalent representations of scientific objects, as presented in Section 4.2.4.
4.3 BIOLOGICAL QUERIES
The design of a BIS strongly depends on how it is going to be used. Section 1.4 of Chapter 1 presents the successive design steps, and Chapter 3 illustrates various design requirements with use cases. Traditional database approaches assume that the relational algebra, or query languages such as the Structured Query Language (SQL) or the Object Query Language (OQL), enable users to express all their queries. Life science shows otherwise. Similar to geographical information systems that aim to let users express complex geometric, topological, or algebraic queries, BIS should enable scientists to express a variety of queries that go beyond the relational algebra. The functionalities required by scientists include sophisticated search mechanisms (see Section 4.3.1) and navigation (see Section 4.3.2), in addition to standard data manipulation. In traditional databases, the semantics of queries are usually bi-valued: true or false. Practice shows that scientists wish to access their data sets through different semantic layers and would benefit from the use of probabilistic or other logical methods to evaluate their queries, as explained in Section 4.3.3. Finally, the complexity of scientific use cases and the applications that support them may drive the design of a BIS to middleware as opposed to a traditional data-driven database approach.
4.3.1 Searching and Mining
Searching typically consists in retrieving entries similar to a given string of characters (phrase, keyword, wildcard, DNA sequence, etc.). In most cases, the data source contains textual documents, and when searching the data source, users expect to retrieve documents that are similar to a given phrase or keyword. Search engines, such as Glimpse6 used to provide search capabilities to GeneCards, use an index to retrieve documents containing the keywords and a ranking system to display ordered retrieved entries. Searching against a sequence data source is performed by honed sequence similarity search engines such as FASTA [37], BLAST [38], and LASSAP [39]. To search sequences, the input string is a sequence, and the ranking of retrieved sequences is customized with a variety of parameters.
Data mining aims to capture patterns out of large data sets with various statistical algorithms. Data mining can be used to discover new knowledge about a data set or to validate an hypothesis. Mining algorithms are often combined with association rules, neural networks, or genetic algorithms. Unlike searching, the data mining approach is not driven by a user’s input expressed through a phrase nor does it apply to a particular data format. Mining a database distinguishes itself from querying a database by the fact that a database query is expressed in a language such as SQL and therefore only captures information organized in the schema. In contrast, a mining tool may exploit information contained in the database that was not organized in the schema and therefore not accessible by a traditional database query.
Most of the query capabilities expected by scientists fall under searching and mining. This was confirmed by the results of a survey of biologists in academia and industry in 2000 [40], where 315 tasks and queries were collected from the answers to the following questions:
1. What tasks do you most perform?
2. What tasks do you commonly perform, that should be easy, but you feel are too difficult?
3. What questions do you commonly ask of information sources and analysis tools?
4. What questions would you like to be able to ask, given that appropriate sources and tools existed, that may not currently exist?
Interestingly, 54% of the collected tasks could be organized into three categories: (1) similarity search, (2) multiple pattern and functional motif search, and (3) sequence retrieval. Therefore, more than half of the identified queries of interest to biologists involve searching or mining capabilities.
Traditional database systems provide SQL as a query language, based on the relational algebra composed of selection (σ), projection (Π), Cartesian product (×), join (![]() ), union (∪), and intersection (∩). These operators perform data manipulation and provide semantics equivalent to that of first order logic [41]. In addition, SQL includes all arithmetic operations, predicates for comparison and string matching, universal and existential quantifiers, summary operations for max/min or count/sum, and GROUP BY and HAVING clauses to partition tables by groups [42]. Commercial database systems extended the query capabilities to a variety of functionalities, such as manipulation of complex datatypes (such as numeric, string, date, time, and interval), OLAP, and limited navigation. However, none of these capabilities can perform the complex tasks specified by biologists in the 2000 survey [40].
), union (∪), and intersection (∩). These operators perform data manipulation and provide semantics equivalent to that of first order logic [41]. In addition, SQL includes all arithmetic operations, predicates for comparison and string matching, universal and existential quantifiers, summary operations for max/min or count/sum, and GROUP BY and HAVING clauses to partition tables by groups [42]. Commercial database systems extended the query capabilities to a variety of functionalities, such as manipulation of complex datatypes (such as numeric, string, date, time, and interval), OLAP, and limited navigation. However, none of these capabilities can perform the complex tasks specified by biologists in the 2000 survey [40].
Other approaches provide search capabilities only, and while failing to support standard data manipulation, they are useful for handling large data sets. They are made available to life scientists as Web interfaces that provide textual search facilities such as GeneCards [9], which uses the powerful Glimpse textual search engine [43], the Sequence Retrieval Service (SRS) [44], and the Entrez interface [45]. GeneCards provides textual search facilities for curated data warehoused in files. SRS, described in detail in Chapter 5, integrates data sources by indexing attributes. It enables queries composed of combinations of textual keywords on most attributes available at integrated databases. Entrez proposes an interesting approach to integrating resources through their similarities. It uses a variety of similarity search tools to index the data sources and facilitate their access through search queries. For instance, the neighbors of a sequence are its homologs, as identified by a similarity score using the BLAST algorithm [38]. On the other hand, the neighbors of a PubMed citation are the articles that use similar terms in their title and abstract [46]. These approaches are limited because they do not allow customized access to the sources. A user looking for PubMed references that have direct protein links will not be able to express the query through Entrez because the interface is designed to retrieve the protein linked from a given citation, not to retrieve all citations linked to a protein. This example, as well as others collected in a 1998 Access article by L. Wong [47], illustrates the weakness of these approaches. A real query language allows customization, whereas a selection of capabilities limits significantly the range of queries biologists are able to ask.
Biologists involve in their queries a variety of search and mining tools and employ traditional data manipulation operators to support their queries. In fact, they often wish to combine them all within a single query. For example, a typical query could start with searching PubMed and only retrieve the references that have direct protein links [47]. Most systems do not support this variety of functionalities yet. Systems such as Kleisli and DiscoveryLink, presented in Chapters 6 and 11, specifically address this issue.
4.3.2 Browsing
Biologists aim to perform complex queries as described in Section 4.3.1, but they also need to browse and navigate the data sets. Systems such as OPM and TAMBIS (see Chapter 7) are designed to provide a user-friendly interface that allows queries through ontologies or object classes, as presented in Section 4.2.5. But they do not provide navigational capabilities that enable access to other scientific objects through a variety of hyperlinks. Web interfaces such as GeneCards offer a large variety of hyperlinks that enable users to navigate directly to other resources such as GenBank, PubMed, and European Molecular Biology Laboratory (EMBL) [34, 48, 76]. Entrez, the Web interface to PubMed, GenBank, and an increasing number of resources hosted at the National Center for Biotechnology Information (NCBI) offer the most sophisticated navigational capabilities. All 15 available resources (as of July 2002) are linked together. For example, a citation in PubMed is linked to related citations in PubMed via the Related Articles, linked to relevant sequences or proteins, respectively, via the Nucleotide Link or the Protein Link. The links are completed by a variety of hyperlinks available in the display of retrieved entries. These navigational capabilities complete the query capabilities and assist the biologists in fulfilling their needs.
The recent development of XML and its navigational capabilities make XPath [49], the language designed to handle navigational queries, and XQuery [50], its extension to traditional data management queries as well as to document queries, good candidates for query languages to manipulate scientific data. There are additional motivations for choosing XML technology to handle scientific data. XML is designed as the standard for data exchange on the Web, and life scientists publish and collect large amounts of data on the Web. In addition, the need for a flexible data representation already evoked the choice of XML in Section 4.2.3. Scientific data providers such as NCBI already offer data in XML format.
Although clearly needed, the development of a navigational foreground for biological data raises complex issues of semantics, as will be presented in Section 4.5.2.
4.3.3 Semantics of Queries
Traditional database approaches use bi-valued semantics: true or false. When a query is evaluated, should any data be missing, the output is NULL. Such semantics are not appropriate for many biological tasks. Indeed, biologists often attempt to collect data with exploring queries, despite missing information. An attribute NULL does not always mean that the value is null but rather that the information is not available yet or is available elsewhere. The rigid semantics of traditional databases may be frustrating for biologists who aim to express queries with different layers of semantics.
Knowledge-based approaches [51] may be used to provide more flexibility. In knowledge bases, reasoning about the possible courses of action replaces the typical database evaluation of a query. Knowledge bases rely on large amounts of expertise expressed through statements, rules, and their associated semantics. Extending BIS with knowledge-based reasoning provides users with customized semantics of queries. BIS can be enhanced by the use of temporal logic that assumes the world to be ordered by time intervals and allows users to reason about time (e.g., “Retrieve all symptoms that occurred before event A”) or fuzzy logic that allows degrees of truth to be attached to statements. Therefore, a solution consists in providing users with a hybrid query language that allows them to express various dependency information, or lack thereof, between events and build a logical reasoning framework on top of such statements of probability. Such an approach has been evaluated for temporal databases [52] and object databases [53]. BIS also could benefit from approaches that would cover the need for addressing object identity as presented in Section 4.2.4, as well as semantic issues such as those to be addressed in Sections 4.4.2 and 4.5.2.
4.3.4 Tool-Driven vs. Data-Driven Integration
Most existing BIS are data-driven: They focus on the access and manipulation of data. But should a BIS really be data-driven? It is not that clear. A traditional database system does not provide any flexibility in the use of additional functionalities. The query language is fixed and does not change. Public or commercial platforms aim to offer integrated software; however, their approach does not provide the ability to integrate easily and freely new softwares as they become available or improve. Commercial integrated platforms also are expensive to use. For scientists with limited budgets, free software is often the only solution.
Some systems provide APIs to use external programs, but the system is no longer the central query processing system; it only processes SQL queries, and an external program executes the whole request. The problem with this approach is that the system, which uses a database system as a component, no longer benefits from the database technology, including efficient query processing (as will be presented in Section 4.4).
Distributed object technology has been developed to cope with the heterogeneous and distributed computing environment that often forces information to be moved from one machine to another, disks to be cross-mounted so different programs can be run on multiple systems, and programs to be re-written in a different programming language to be compiled and executed on another architecture. The variety of scientific technology presented in Section 4.1.2 often generates significant waste of time and resources. Distributed object technology includes Common Object Request Broker Architecture (CORBA), Microsoft Distributed Component Object Model (DCOM), and Java Remote Method Invocation (RMI). This technology is tools-driven and favors a computational architecture that interoperates efficiently and robustly. Unlike traditional databases, it allows flexible access to computational resources with easy registration and removal of tools. For these reasons, many developers of BIS are currently using this technology. For sake of efficiency, a new version of TAMBIS no longer uses CPL but provides a user-friendly ontology of biological data sources using CORBA clients to retrieve information from these sources [54] (see Chapter 5). CORBA appears to be suitable for creating wrappers via client code generation from interface definition language (IDL) definitions. The European Bioinformatics Institute (EBI) is leading the effort to make its data sources CORBA compliant [48, 55]. Unfortunately, most data providers do not agree with this effort. Concurrent to the CORBA effort, NCBI, the American institute, and EBI provide their data sources in XML format.
A lot of interest has been given to grid architectures. A grid architecture aims to enable computing as well as data resources to be delivered and accessed seamlessly, transparently, and dynamically, when needed, on the Internet. The name grid was inspired by the electricity power grid. A biologist should be able to plug into the grid like an appliance is plugged into an outlet and use resources available on the grid transparently. The grid is an approach to a new generation of BIS. Examples of grids include the Open Grid Services Architecture (OGSA) [56] and the open source problem-solving environment Cactus [57]. TeraGrid [58] and Data Grid [59] are international efforts to build grids.
These tool-driven proposals do not yet solve the many problems of resource selection, query planning, optimization, and other semantics issues as will be presented in Sections 4.4 and 4.5.
4.4 QUERY PROCESSING
In the past, query processing often received less attention from designers of BIS. Indeed, BIS developers devoted most of their effort to meeting the needs for integration of data sets and applications and providing a user-friendly interface for scientists. However, as the data sets get larger, the applications more time-consuming, and the queries more complex, the specification for fast query processing becomes critical.
4.4.1 Biological Resources
A BIS must adequately capture and exploit the diverse, and often complex, query processing, or other computational capabilities of biological resources by specifying them in a catalog and using them at both query formulation and query evaluation. The W3C Semantic Web Activity [60, 61] aims to provide a meta-data layer to permit people and applications to share data on the Web. Recent efforts within the bioinformatics community address the use of OIL [62] to capture alternative representations of data to extend biomolecular ontologies [63]. Such efforts focusing on data representation of the contents of the sources must be extended to capture meta-data along several dimensions, including (1) the coverage of the information sources, (2) the capabilities, links, and statistical patterns, (3) the data delivery patterns of the resources, and (4) data representation and organization at the source.
The coverage of information sources is useful in solving the so-called source relevance problem, which involves deciding which of the myriad sources are relevant for the user and to evaluate the submitted query. Directions to characterize and exploit coverage of information sources include local, closed world assumptions, which state that the source is complete for a specific part of the database [64, 65]; quantifications of coverage (e.g., the database contains at least 90% of the sequences), or intersource overlaps (e.g., the EMBL Nucleotide Sequence Database has a 75% likely overlap with DDBJ for sequences annotated with “calcium channel”) [66–68]. Characterizing coverage enables the exploitation of coverage positioning of data sources from complement to partial or complete overlap (mirror sites).
Source capabilities capture the types of queries supported by the sources, the access pattern limitations, the ability to handle limited disjunction, and so on. Although previous research has addressed capabilities [69–74], it has not addressed the diverse and complex capabilities of biological sources. Recent work aims to identify the properties of sophisticated source capabilities including text search engines, similarity sequence search engines such as BLAST, and multiple sequence alignment tools such as Cluster [75, 76]. Their characteristics are significantly more complex than the capabilities addressed up to now, and their use is dramatically costly in terms of processing time. In addition, many of the tools are closely coupled to the underlying source, which requires the simultaneous identification of capabilities and coverage.
Statistical patterns include the description of information clusters and the selectivities of all or some of the data access mechanisms and capabilities. The use of simple statistical patterns has been studied [65, 66, 77]. However, BIS should exploit statistical patterns of real data sources that are large, complex, and constantly evolving (as opposed to their simplified simulations).
Delivery patterns include the response time, that is, units of time needed to receive the first block of answers, the size of these blocks and so on. Delivery patterns may affect the query evaluation process significantly. Depending on the availability of the proper indices, a source may either return answers in decreasing order of matching (from best to worst) or in an arbitrary (unordered) manner. Other delivery profiles include whether information can be provided in a sorted manner for certain attributes or not. These types of profiles will be essential in identifying sources to get the first, best answers. This is useful when a user expects to get the answers to a query sorted in a pre-defined relevant order (the more relevant answers are the first returned). Delivery patterns can also be exploited to provide users with a faster, relevant, but maybe incomplete answer to a query. BIS should exploit delivery patterns in conjunction with the actual capabilities supported by the sources.
The access and exploitation of the previously mentioned meta-knowledge of biological resources offers several advantages. First, it enables the comparison of diverse ways to evaluate a query as explained in the next section. Further, it can characterize the most efficient way to evaluate a query, as will be presented in Section 4.4.3.
4.4.2 Query Planning
Query planning consists in considering the many potential combinations of accesses to evaluate a query. Each combination is a query evaluation plan. Consider the query (Q) defined as follows:
(Q)“Return accession numbers and definitions of GenBank EST sequences that are similar (60% identical over 50AA) to ‘Calcium channel’ sequences in Swiss-Prot that have references published since 1995 and mention ‘brain.’” [78]
There exist many plans to evaluate the query. One possible plan for this query is illustrated in Figure 4.1 and described as follows: (1) access PubMed and retrieve references published since 1995 that mention brain; (2) extract from all these references the Swiss-Prot identifiers; (3) obtain the corresponding sequences from Swiss-Prot whose function is calcium channel; and (4) execute a BLAST search using a wrapped BLAST application to retrieve similar sequences from GenBank (gbest sequences).

FIGURE 4.1 First plan for evaluating query (Q) [75].
Figure 4.2 presents an alternative approach that first accesses Swiss-Prot and retrieves sequences whose function is calcium channel. In parallel, it retrieves the citations from PubMed that mention brain and are published since 1995 and extracts sequences from them. Then it determines which sequences are in common. Finally, it executes a BLAST search to retrieve similar sequences from GenBank (gbest sequences).
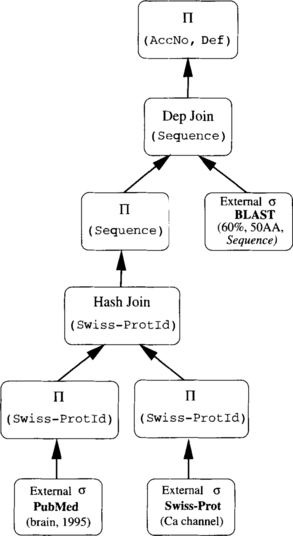
FIGURE 4.2 Second plan for evaluating query (Q) [75].
Scientific resources overlap significantly. The variety of capabilities, as well as the coverages and statistical patterns presented in Section 4.4.1, offer many alternative evaluation plans for a query. The number of evaluation plans is exponential to the size of similar resources. Therefore not all plans should be evaluated to answer a query. To select the plan to evaluate a given query, first the semantics of the plan should be captured accurately. Indeed, two plans may be similar and yet not semantically equivalent. For example, suppose a user is interested in retrieving the sequences relevant to the article entitled “Suppression of Apoptosis in Mammalian Cells by NAIP and a Related Family of IAP Genes” published in Nature and referenced by 8552191 in PubMed. A first plan is to extract the GenBank identifiers explicitly provided in the MEDLINE format of the reference. A second plan consists in using the capability Nucleotide Link, provided at NCBI. The two plans are not semantically equivalent because the first plan returns four GenBank identifiers when the Nucleotide Link returns eight GenBank identifiers (as of August 2001).
Verifying whether two plans are semantically equivalent, that is, if the answers that are returned from the two plans are identical, is non-trivial and depends on the meta-data of the particular resources used in each plan. This issue is closely related to navigation over linked resources as will be presented in Section 4.5.2. In addition, two semantically equivalent plans may differ dramatically in terms of efficiency, as is explained in the next section.
4.4.3 Query Optimization
Query optimization [79, 80] is the science and the art of applying equivalence rules to rewrite the tree of operators evoked in a query and produce an optimal plan. A plan is optimal if it returns the answer in the least time or using the least space. There are well known syntactic, logical, and semantic equivalence rules used during optimization [79]. These rules can be used to select an optimal plan among semantically equivalent plans by associating a cost with each plan and selecting the lowest overall cost. The cost associated with each plan is generated using accurate metrics such as the cardinality or the number of result tuples in the output of each operator, the cost of accessing a source and obtaining results from that source, and so on. One must also have a cost formula that can calculate the processing cost for each implementation of each operator. The overall cost is typically defined as the total time needed to evaluate the query and obtain all of the answers.
The characterization of an optimal, low-cost plan is a difficult task. The complexity of producing an optimal, low-cost plan for a relational query is NP-complete [79–81]. However, many efforts have produced reasonable heuristics to solve this problem. Both dynamic programming and randomized optimization based on simulated annealing provide good solutions [82–84].
A BIS could be improved significantly by exploiting the traditional database technology for optimization extended to capture the complex metrics presented in Section 4.4.1. Many of the systems presented in this book address optimization at different levels. K2 (see Chapter 8 Section 8.1) uses rewriting rules and a cost model. P/FDM (see Chapter 9) combines traditional optimization strategies, such as query rewriting and selection of the best execution plan, with a query-shipping approach. DiscoveryLink (see Chapter 11) performs two types of optimization: query rewriting followed by a cost-based optimization plan. KIND (see Chapter 12) is addressing the use of domain knowledge into executable meta-data. The knowledge of biological resources can be used to identify the best plan with query (Q) defined in Section 4.4.2 as illustrated in the following.
The two possible plans illustrated in Figures 4.1 and 4.2 do not have the same cost. Evaluation costs depend on factors including the number of accesses to each data source, the size (cardinality) of each relation or data source involved in the query, the number of results returned or the selectivity of the query, the number of queries that are submitted to the sources, and the order of accessing sources.
Each access to a data source retrieves many documents that need to be parsed. Each object returned may generate further accesses to (other) sources. Web accesses are costly and should be as limited as possible. A plan that limits the number of accesses is likely to have a lower cost. Early selection is likely to limit the number of accesses. For example, the call to PubMed in the plan illustrated in Figure 4.1 retrieves 81,840 citations, whereas the call to GenBank in the plan in Figure 4.2 retrieves 1616 sequences. (Note that the statistics and results cited in this paper were gathered between April 2001 and April 2002 and may no longer be up to date.) If each of the retrieved documents (from PubMed or GenBank) generated an additional access to the second source, clearly the second plan has the potential to be much less expensive when compared to the first plan.
The size of the data sources involved in the query may also affect the cost of the evaluation plan. As of May 4, 2001, Swiss-Prot contained 95,674 entries whereas PubMed contained more than 11 million citations; these are the values of cardinality for the corresponding relations. A query submitted to PubMed (as used in the first plan) retrieves 727,545 references that mention brain, whereas it retrieves 206,317 references that mention brain and were published since 1995. This is the selectivity of the query. In contrast, the query submitted to Swiss-Prot in the second plan returns 126 proteins annotated with calcium channel.
In addition to the previously mentioned characteristics of the resources, the order of accessing sources and the use of different capabilities of sources also affects the total cost of the plan. The first plan accesses PubMed and extracts values for identifiers of records in Swiss-Prot from the results. It then passes these values to the query on Swiss-Prot via the join operator. To pass each value, the plan may have to send multiple calls to the Swiss-Prot source, one for each value, and this can be expensive. However, by passing these values of identifiers to Swiss-Prot, the Swiss-Prot source has the potential to constrain the query, and this could reduce the number of results returned from Swiss-Prot. On the other hand, the second plan submits queries in parallel to both PubMed and Swiss-Prot. It does not pass values of identifiers of Swiss-Prot records to Swiss-Prot; consequently, more results may be returned from Swiss-Prot. The results from both PubMed and Swiss-Prot have to be processed (joined) locally, and this could be computationally expensive. Recall that for this plan, 206,317 PubMed references and 126 proteins from Swiss-Prot are processed locally. However, the advantage is that a single query has been submitted to Swiss-Prot in the second plan. Also, both sources are accessed in parallel.
Although it has not been described previously, there is a third plan that should be considered for this query. This plan would first retrieve those proteins annotated with calcium channel from Swiss-Prot and extract MEDLINE identifiers from these records. It would then pass these identifiers to PubMed and restrict the results to those matching the keyword brain. In this particular case, this third plan has the potential to be the least costly. It submits one sub-query to Swiss-Prot, and it will not download 206,317 PubMed references. Finally, it will not join 206,317 PubMed references and 126 proteins from Swiss-Prot locally.
Optimization has an immediate impact in the overall performance of the system. The consequences of the inefficiency of a system to execute users’ queries may affect the satisfaction of users as well as the capabilities of the system to return any output to the user. These issues are presented in Chapter 13.
4.5 VISUALIZATION
An important issue when designing a BIS is visualization. Scientific data are available in a variety of media, and life scientists expect to access all these data sets by browsing through correspondences of interest, regardless of the medium or the resource used. The ability to combine and visualize data is critical to scientific discovery. For example, KIND presented in Chapter 12 provides several visual interfaces to allow users to access and annotate the data. For example, the spatial annotation tool displays 2D maps of brain slices when another interface shows the UMLS concept space.
4.5.1 Multimedia Data
Scientific data are multimedia; therefore, a BIS should be designed to manage images, pathways, maps, 3D structures, and so on regardless of their various formats (e.g., raster, bitmap, GIF, TIFF, PCX). An example of the variety of data formats and media generated within a single application is illustrated in Chapter 10. Managing multimedia data is known to be a difficult task. A multimedia management system must provide uniform access transparent to the medium or format. Designing a multimedia BIS raises new challenges because of the complexity and variety of scientific queries. The querying process is an intrinsic part of scientific discovery. A BIS user’s interface should enable scientists to visualize the data in an intuitive way and access and query through this representation. Not only do scientists need to retrieve data in different media (e.g., images), but they also need the ability to browse the data with maps, pathways, and hypertext. This means a BIS needs to express a variety of relationships among scientific objects. The difficulties mentioned previously regarding the identification of scientific objects (see Section 4.2.4) are dramatically increased by the need to capture the hierarchy of relationships. Scientific objects such as genes, proteins, and sequences can be seen as classes in an entity-relationship (ER) model; and a map can be seen as the visualization of a complex ER diagram composed of many classes, isa relationships, relationships, and attributes. Each class can be populated by data collected from different data sources and the relationships corresponding to different source capabilities. For example, two classes, gene and publication, can be respectively populated with data from GeneCards and PubMed. The relationship from the class gene to the class publication, expressing the publications in which the gene was published, can be implemented by capturing the capability available at GeneCards that lists all publications associated with a gene and provides their PubMed identifiers. The integration data schema is very complex because data and relationships must be integrated at different levels of the nested hierarchy. Geographical information systems address similar issues by representing maps at different granularities, encompassing a variety of information.
Many systems have been developed to manage geographical and spatial data, medical data, and multimedia data. Refer to Spatial Databases—with Applications to GIS [85], Neural Networks and Artifical Intelligence for Biomedical Engineering, [86] and Principles of Multimedia Database Systems [87] for more information. However, very little has been done to develop a system to integrate scientific multimedia systems seamlessly. The following section partially addresses the problem by focusing on relationships between scientific objects.
4.5.2 Browsing Scientific Objects
Scientific entities are related to each other. A gene comprises one or more sequences. A protein is the result of a transcription of dioxyribonucleic acid (DNA) into RNA followed by a translation. Sequences, genes, and proteins are related to reference publications. These relationships are often represented by links (and hyperlinks). For example, there is a relationship between an instance of a gene and instances of the set of sequences that comprise the gene.
The attributes describing an entity, the relationship associating the entity to other entities, and most importantly, the semantics of the relationships, correspond to the complete functional characterization of an entity. Such a characterization, from multiple sources and representing multiple points of view, typically introduces discrepancies. Examples of such discrepancies include dissimilar concepts (GenBank is sequence-centric whereas GeneCards is gene-centric), dissimilar attribute names (the primary GeneCards site has an attribute protein whereas a mirror represents the same information as an attribute product), and dissimilar values or properties (the gene TP53 is linked to a single citation in the data source HUGO, 35 citations in the data source GDB, and two citations in the data source GeneCards).
A BIS integrating multiple data sources should allow life scientists to browse the data over the links representing the relationships between scientific objects. A path is a sequence of classes, starting and ending at a class and intertwined with links. Two paths with identical starting and ending classes may be equivalent if they have the same semantics. The resolution of the equivalence of paths is also critical to developing efficient systems as discussed in Section 4.4.3. Semantic equivalence is a difficult problem, as illustrated in the following example. Consider a link from PubMed citations to sequences in GenBank. This link can be physically implemented in two different ways: (1) by extracting GenBank identifiers from the MEDLINE format of the PubMed citation, or (2) by capturing the Nucleotide Link as implemented via the Entrez interface. Both implementations expect to capture all the GenBank identifiers relevant to a given PubMed citation. These two links have same starting and ending classes; however, they do not appear to be equivalent. Using the first implementation, the PubMed citation 8552191 refers to four GenBank identifiers. In contrast, the Nucleotide Link representing the second property returns eight GenBank identifiers. Based on the dissimilar cardinality of results (the number of returned sequences), the two properties are not identical. This can also be true for paths (informal sequences of links) between entities. To make the scenario more complex, there could be multiple alternate paths (links) between a start entity and an end entity implemented in completely different sources.
A BIS able to exploit source capabilities and information on the semantics of the relationships between scientific objects would provide users the ability to browse scientific data in a transparent and intuitive way.
4.6 CONCLUSION
Traditional technology often does not meet the needs of life scientists. Each research laboratory uses significant manpower to adjust and customize as much as possible the available technology. Because of the failure of traditional approaches to support scientific discovery, life scientists have proven to be highly creative in developing their own tools and systems to meet their needs. Traditional database systems lack flexibility: Life scientists use flat files instead. Some of the tools and systems developed in scientific laboratories may not meet the expectations of computer scientists, but they perform and support thousands of life scientists. The development of BIS is driven by the needs of a community. But practice shows that the community now needs the development of systems that are more engineered than before, and computer scientists should be involved.
There are good reasons for traditional technology to fail to meet the requirements of BIS. Databases are data-driven and lack flexibility at the level of data representation. XML and other semi-structured approaches may offer this needed flexibility, but the development of native semi-structured systems still is in its infancy. Knowledge bases offer different semantic layers to leverage queries with the exploration process, but they should be coupled with databases to perform traditional data manipulation. On the other hand, agent architectures and grids provide flexible and transparent management of tools. Each of these approaches may and should contribute to the design of BIS.
The systems presented in this book constitute the first generation of BIS. Each system addresses some of the requirements presented in Chapter 2. Each presented system still is successfully used by life scientists; however, the development of each of these systems told a lesson. To be successful, the design of the next generation of BIS should take advantage of these lessons and exploit and combine all existing approaches.
ACKNOWLEDGMENTS
The author wishes to thank Louiqa Raschid and Barbara Eckman for fruitful discussions that contributed to some of the material presented in this chapter.
REFERENCES
[1] Baxevanis, A. The Molecular Biology Database Collection: 2002 Update. Nucleic Acids Research. 2002;30(no. 1):1–12.
[2] Burks, C., Molecular Biology Database List. Nucleic Acids Research. 1999;27(no. 1):1–9. http://nar.oupjournals.Org/cgi/content/full/27/l/l
[3] Baxevanis, A. The Molecular Biology Database Collection: An Online Compilation of Relevant Database Resources. Nucleic Acids Research. 2000;28(no. 1):1–7.
[4] Baxevanis, A. The Molecular Biology Database Collection: An Updated Compilation of Biological Database Resources. Nucleic Acids Research. 2001;29(no. 1):1–10.
[5] U.S.Department of Energy Human Genome Program. Report from the 1999 U. S. DOE Genome Meeting: Informatics. Human Genome News. 1999;10(no.3–4):8.
[6] Januaryhttp://www.ncbi.nlm.nih.gov/Genbank. Benson, D., Karsch-Mizrachi, I., Lipman, D., et al. GenBank. Nucleic Acids Research. 2000;28(no. 1):15–18.
[7] Januaryhttp://www.expasy.ch/sprot. Bairoch, A., Apweiler, R. The SWISS-PROT Protein Sequence Databank and Its Supplement TrEMBL in 1999. Nucleic Acids Research. 1999;27(no. 1):49–54.
[8] Saccharomyces Genome Database (SGD). http://genome-www.Stanford.edu/saccaromyces/. Department of Genetics, Stanford University.
[9] Julyhttp://bioinformatics.weizmann.ac.il/cards/CABIOS/paper.html. Rebhan, M., Chalifa-Caspi, V., Prilusky, J., et al. GeneCards: A Novel Functional Genomics Compendium with Automated Data Mining And Query Reformulation Support. Bioinformatics. 1998;14(no. 8):656–664.
[10] PubMed. http://www.ncbi.nlm.gov/pubmed/. National Library of Medicine.
[11] revised March 12 Gen, Bank, Growth of GenBank. NCBI GenBank Statistics 2002. http://www.ncbi.nlm.nih.gov/Genbank/genbankstats.html
[12] February International Human Genome Sequencing Consortium. Initial Sequencing and Analysis of the Human Genome. Nature. 2001;409:860–921.
[13] February Venter, J., Adams, M., Myers, E., et al. The Sequence of the Human Genome. Science. 2001;291(no. 5507):1304–1351.
[14] Croft, L., Schandorff, S., Clark, F., et al. Isis: The Intron Information System Reveals the High Frequency of Alternative Splicing in the Human Genome. Nature Genetics. 2000;24:340–341.
[15] Graveley, B. Alternative Splicing: Increasing Diversity in the Proteomic World. Trends in Genetics. 2001;17(no. 2):100–107.
[16] no. 132 Misener, S., Krawetz, S. Bioinformatics: Methods and Protocols. In: Methods in Molecular Biology. Totowa, NJ: Humana Press; 1999.
[17] Hollingsworth, D., The Workflow Reference Model. Hampshire, UK: Workflow Management Coalition; 1995. http://www.wfmc.org/standards/docs/tc003vll.pdf
[18] Buneman, P., Davidson, S., Hillebrand, G., et al, A Query Language and Optimization Techniques for Unstructured Data, Proceedings of the 1996 ACM SIGMOD International Conference on Management of Data. Montreal, June 4–6, 1996. New York: ACM Press; 1996:505–516.
[19] Nestorov, S., Ullman, J., Wiener, J., et al, Representative Objects: Concise Representations of Semi-Structured Hierarchical Data, Proceedings of the Thirteenth International Conference on Data Engineering. April 7–11, 1997 Birmingham U.K. Washington, D.C: IEEE Computer Society; 1997:79–90.
[20] Fan, W., Path Constraints for Databases with or without Schemas, Ph.D. dissertation. University of Pennsylvania; 1999.
[21] May 2 Fallside, D. XML Schema Part 0: Primer: World Wide Web Consortium (W3C) Recommendation. 2001. http://www.w3c.org/TR/2001/REC
[22] May 2 Thompson, H., Beech, D., Maloney, M., et al. XML Schema Part 1: Structures: World Wide Web Consortium (W3C) Recommendation. 2001. http://www.w3c.org/TR/2001/REC
[23] May 2 Biron, P., Malhotra, A. XML Schema Part 2: Datatypes: World Wide Web Consortium (W3C) Recommendation. 2001. http://www.w3.org/TR/2001/REC
[24] Abiteboul, S., Buneman, P., Suciu, D. Data on the Web. San Francisco: Morgan Kaufmann; 2000.
[25] April Wain, H., Bruford, E., Lovering, R., et al. Guidelines for Human Gene Nomenclature (2002). Genomics. 2002;79(no. 4):464–470.
[26] Human Genome Database (GDB). http://www.gdb.org. The Hospital for Sick Children, Baltimore, MD: Johns Hopkins University.
[27] October Frezal, J. GenAtlas Database, Genes and Development Defects. Comptes Rendus de VAcadémie des Sciences-Series III: Sciences de la Vie. 1998;321(no. 10):805–817.
[28] GenAtlas. Direction des Systèmes d’Information, Université Paris 5, France. http://www.dsi.univ-paris5.fr/genatlas/.
[29] Hamosh, A., Scott, A.F., Amberger, J., et al. Online Mendelian Inheritance in Man (OMIM). Human Mutation. 2000;15(no. 1):57–61.
[30] Online Mendelian Inheritance in Man (OMIM). http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=omim. World Wide Web interface developed by the National Center for Biotechnology information (NCBI, National Library of Medicine.
[31] Pruitt, K., Katz, K., Sciotte, H., et al. Introducing Refseq and LocusLink: Curated Human Genome Resources at the NCBI. Trends in Genetics. 2000;16(no. 1):44–47.
[32] LocusLink. http://www.ncbi.nlm.nih.gov/locuslink/. National Center for Biotechnology Information (NCBI), National Library of Medicine.
[33] Schuler, G.D. Pieces of the Puzzle: Expressed Sequence Tags and the Catalog of Human Genes. Journal of Molecular Medicine. 1997;75(no. 10):694–698.
[34] Chen, I.M.A., Markowitz, V.M. An Overview of the Object-Protocol Model (OPM) and OPM Data Management Tools. Information Systems. 1995;20(no. 5):393–418.
[35] Berlin, J., Motro, A., Autoplex: Automated Discovery of Content for Virtual Databases, Proceedings of the 9th International Conference on Cooperative Information Systems. Trento, Italy, September 5–7, 2001. New York: Springer; 2001:108–122.
[36] Doan, A., Domingos, P., Levy, A., Learning Source Descriptions for Data Integration, Proceedings of the Third International Workshop on the Web and Databases. Dallas, May 18–19, 2000. 2000:81–86.
[37] April Pearson, W., Lipman, D. Improved Tools for Biological Sequence Comparison. Proceedings of the National Academy of Science. 1988;85(no. 8):2444–2448.
[38] Octoberhttp://www.ncbi.nlm.nih.gov/BLAST. Altschul, S., Gish, W., Miller, W., et al. Basic Local Alignment Search Tool. Journal of Molecular Biology. 1990;215(no. 3):403–410.
[39] Glemet, E., Codani, J-J., LASSAP: A LArge Scale Sequence CompArison Package. Bioinformatics. 1997;13(no. 2):137–143. www.gene.it.com
[40] Stevens, R.D., Goble, C.A., Baker, P., et al. A Classification of Tasks in Bioinformatics. Bioinformatics. 2001;17(no. 2):180–188.
[41] Abiteboul, S., Hull, R., Vianu, V. Foundations of Databases. Boston: Addison-Wesley; 1995.
[42] June 2 National Institute of Standards Technology. Database Language SQL. 1993. http://www.itl.nist.gov/fipspubs/fipl27.2.htm
[43] Manber, U., Sun, W. GLIMPSE: A Tool to Search Through Entire File Systems. In: Proceedings of USENIX Conference. Berkeley, CA: USENIX Association; 1994:23–32.
[44] Etzold, T., Argos, P., SRS: An Indexing and Retrieval Tool for Flat File Data Libraries. Computer Applications of Biosciences. 1993;9(no. 1):49–57. http://srs.ebi.ac.uk
[45] Schuler, G., Epstein, J., Ohkawa, H., et al. Entrez: Molecular Biology Database and Retrieval System. Methods in Enzymology. 1996;266:141–162.
[46] Wilbur, W., Yang, Y. An Analysis of Statistical Term Strength and Its Use in the Indexing and Retrieval of Molecular Biology Texts. Computers in Biology and Medicine. 1996;26(no. 3):209–222.
[47] June Wong, L. Some MEDLINE Queries Powered by Kleisli. ACCESS. 1998;25:8–9.
[48] Stoesser, G., Baker, W., Van Den Broek, A., et al. The EMBL Nucleotide Sequence Database: Major New Developments. Nucleic Acids Research. 2003;31(no. 1):17–22.
[49] November 16 Clark, J., DeRose, S. XML Path Language (XPath): World Wide Web Consortium (W3C) Recommendation. 1999. http://www.w3.org/TR/xpath
[50] Chamberlin, D., Florescu, D., Robie, J., et al. XQuery: A Query Language for XML: World Wide Web Consortium (W3C) Recommendation. 2000. http://www.w3.org/TR/xmlquery
[51] Russel, S., Norvig, P. Artificial Intelligence: A Modern Approach. Upper Saddle River, NJ: Prentice Hall; 1995.
[52] Kraus, S., Dix, J., Subrahmanian, V.S. Probabilistic Temporal Databases. Artificial Intelligence Journal. 2001;127(no. 1):87–135.
[53] Eiter, T., Lu, J., Lucasiewicz, T., et al. Probabilistic Object Bases. ACM Transactions on Database Systems. 2001;26(no. 3):264–312.
[54] Stevens, R., Brass, A., Using CORBA Clients in Bioinformatics Applications. Collaborative Computational Project 11 Newsletter 1998;2.1(no. 3). http://www.hgmp.mrc.ac.uk/CCPl1/CCP1lnewsletters/CCPllNewsletterIssue3.pdf
[55] Wang, L., Rodriguez-Tomé, P., Redaschi, N., et al. Accessing and Distributing EMBL Data Using CORBA (Common Object Request Broker Architecture). Genome Biology. 1(no. 5), 2000.
[56] Globus. Open grid services architecture (OGSA). http://www.Globus.org/ogsa/.
[57] Cactus: open source problem solving environment, http://www.cactuscode.org/.
[58] NSF funded. Teragrid. http://www.teragrid.org/.
[59] European Union. Datagrid. http://eu-datagrid.web.cern.ch/eu-datagrid/.
[60] June Berners-Lee, T., Connoly, D., Swick, R., Web Architecture: Describing and Exchanging Data. W3C Note 1999. http://www.w3.org/1999/04/WebData
[61] May Berners-Lee, T., Hendler, J., Lassila, O. The Semantic Web. Scientific American. 2001.
[62] Fensel, D., Horrocks, I., van Harmelem, R., et al, OIL in a Nutshell, Proceedings of the 12th International European Knowledge Acquisition Conference, EKAW2000. Juan-les-Pins, France, October 2–6, 2000. LNAI: Springer Verlag; 2000.
[63] Critchlow, T. Report on XEWA-00: The XML-Enabled Wide-Area Searches for Bioinformatics Workshop. New York: IEEE Computer Society; 2000.
[64] Friedman, M., Weld, D., Efficiently Executing Information-Gathering Plans, Proceedings of the Fifteenth International Joint Conference on Artificial Intelligence. Nagoya, Japan, August 23–29, 1997. San Francisco: Morgan Kaufmann; 1997:785–791.
[65] Lambrecht, E., Kambhampati, S., Gnanaprakasam, S., Optimizing Recursive Information-Gathering Plans, Proceedings of the Sixteenth International Joint Conference on Artificial Intelligence. Stockholm, July 31–August 6, 1999. San Francisco: Morgan Kaufmann; 1999:1204–1211.
[66] Florescu, D., Koller, D., Levy, A.Y., Using Probabilistic Information in Data Integration, Proceedings of 23rd International Conference on Very Large Data Bases. August 25–29, 1997, Athens. San Francisco: Morgan Kaufmann; 1997:216–225.
[67] conjunction with the ACM SIGMOD Mihaila, G., Raschid, L., Vidal, M-E., Using Quality of Data Metadata for Source Selection and Ranking, Proceedings of the Third International Workshop on the Web and Databases. Dallas, May 18–19, 2000. 2000:93–98.
[68] conjunction with ACM PODS/SIGMOD Mihaila, G.A., Raschid, L., Vidal, M.-E., Using Quality of Data Metadata for Source Selection and Ranking. Informal proceedings. Suciu D., Vossen G., eds. Proceedings of the Third International Workshop on the Web and Databases, WebDB 2000. Dallas, Texas, May 18–19, 2000. 2000.
[69] Baru, C., Gupta, A., Ludäscher, B., et al, XML-Based Information Mediation with MIX, Proceedings ACM SIGMOD International Conference on Management of Data. June 1–3, 1999, Philadelphia. New York: Association for Computing Machinery(ACM) Press; 1999:597–599.
[70] Levy, A., Rajaraman, A., Ordille, J., Querying Heterogeneous Information Sources Using Source Descriptions, Proceedings of 22nd International Conference on Very Large Data Bases. September 3–6, 1996, Mumbai, India. San Francisco: Morgan Kaufmann; 1996:251–262.
[71] Papakonstantinou, Y., Vassalos, V., Query Rewriting for Semistructured Data, Proceedings of the ACM SIGMOD International Conference on Management of Data. June 1–3, 1999, Philadelphia. New York: ACM Press; 1999:455–466.
[72] Vidal, M.E., Mediation Techniques for Multiple Autonomous Distributed Information Sources, Ph.D. dissertation. Caracas, Venezuela: Universidad Simon Bolivar; 2000.
[73] Vassalos, V., Papakonstantinou, Y., Describing and Using Query Capabilities of Heterogeneous Sources, Proceedings of 23rd International Conference on Very Large Data Bases. August 25–29, 1997, Athens. San Francisco: Morgan Kaufmann; 1997:256–265.
[74] Yemeni, R., Li, C., Garcia-Molina, H., et al, Computing Capabilities of Mediators, Proceedings ACM SIGMOD International Conference on Management of Data. June 1–3, 1999, Philadelphia. New York: ACM Press; 1999:443–454.
[75] Eckman, B., Lacroix, Z., Raschid, L., Optimized Seamless Integration of Biomolecular Data, 2nd IEEE International Symposium on Bioinformatics and Bio engineering. Bethesda, Maryland, November 4–5, 2001. Washington D.C.: IEEE Computer Society, D.C.; 2001:23–32.
[76] June Lacroix, Z. Biological Data Integration: Wrapping Data and Tools. IEEE Transactions on Information Technology in Biomedicine. 2002;6(no. 2):123–128.
[77] Nie, Z., Kambhampati, S., Joint Optimization of Cost and Coverage of Query Plans in Data Integration, Proceedings of the 2001 ACM CIKM International Conference on Information and Knowledge Management. November 5–10, 2001, Atlanta. New York: ACM Press; 2001:223–230.
[78] Eckman, B., Kosky, A., Laroco, L. Extending Traditional Query-Based Integration Approaches for Functional Characterization of Post-Genomic Data. Bioinformatics. 2001;17(no. 7):587–601.
[79] Ullman, J. Principles of Database and Knowledge-Base Systems. Palo Alto, CA: Computer Science Press; 1989;volume II.
[80] Ullman, J. Information Integration Using Logical Views. In: Proceedings of the Sixth International Conference on Database Theory. Springer; 1997:19–40.
[81] Morris, K., An Algorithm for Ordering Subgoals in NAIL, Proceedings of the Seventh ACM SIGACT-SIGMOD-SIGART Symposium on Principles of Database Systems. March 21–23, 1988, Austin, Texas. New York: ACM Press; 1988:82–88.
[82] Ioanidis, Y., Kang, Y., Randomized Algorithms for Optimizing Large Join Queries, Proceedings of the 1990 ACM SIGMOD International Conference on Management of Data. Atlantic City, N], May 23–25, 1990. New York: ACM Press; 1990:312–321.
[83] Selinger, P., Astrahan, M., Chamberlin, D., et al, Access Path Selection in a Relational Database Management System, Proceedings of the 1979 ACM SIGMOD International Conference on Management of Data. Boston, May 30-Junel. New York: ACM Press; 1979:23–34.
[84] Steinbrunn, M., Moerkotte, G., Kemper, A. Heuristic and Randomized Optimization for the Join Ordering Problem. VLDB Journal. 1997;6(no. 3):191–208.
[85] Rigaux, P., Scholl, M., Voisard, A. Spatial Databases—With Applications to GIS. San Francisco: Morgan Kaufmann; 2001.
[86] Hudson, D., Cohen, M. Neural Networks and Artificial Intelligences for Biomedical Engineering. New York: IEEE Press; 2000.
[87] Subrahmanian, V.S. Principles of Multimedia Database Systems. San Francisco: Morgan Kaufmann; 1998.
1Oracle9i was developed by the Oracle corporation (see http://www.oracle.com).
2SQL Server 2000 is a product of the Microsoft Corporation (see http://www.microsoft.com).
3Tamino XML server is a commercial XML management system from SoftwareAG.
4ToX is an academic XML management system being developed at the University of Toronto.
5Galax was developed at the Bell Laboratory of Lucent Technology (see http://www.db.bell-labs.com/galax).
6The Glimpse search engine was developed and is maintained at the University of Arizona and is available at http://glimpse.cs.arizona.edu/.