
Chapter 3
Computational Approaches for Predicting hERG Activity
Vinicius M. Alves, Rodolpho C. Braga and Carolina Horta Andrade
LabMol – Laboratory for Molecular Modeling and Design, Faculty of Pharmacy, Federal University of Goias, Goiania, GO, Brazil
Chapter Menu
3.1 Introduction
The human ether-à-go-go related gene (hERG) is a gene (kcnh2) that encodes for a protein named Kv11.1, the α subunit of potassium ion channels. The hERG channels are expressed in a variety of tissues, but its clinical significance is more understood in the heart. This voltage-gated channel contributes to the coordination of the heart beat by mediating the repolarization of the cardiac action potential [1]. The structure and function of the heart are commonly evaluated by performing electrocardiography. In this examination, the electrical activity of the heart is recorded, where the QT interval is the measure of the duration of ventricular depolarization and repolarization in an electrocardiogram [2].
Abnormalities in the structure of hERG caused by different mutations are associated with increase or decrease of the QT interval, both related to sudden death. Short QT syndrome is a very rare condition caused by mutation and it is not discussed in this chapter. The long QT syndrome, also termed Torsade de Pointes, may be caused by genetic defects or induced by drugs [3]. Chemically induced prolongation of the QT interval is caused by the blockage of hERG and this is an important clinical issue that is a point of focus of current drug development and regulation [4, 5].
The hERG channel earned notoriety for being associated with potentially fatal cardiac arrhythmia [6]. According to the WITHDRAWN database [7], twenty-seven drugs have been withdrawn from the market because of their blockage of hERG channels, including blockbusters [8] such as astemizole, terfenadine [9], cisapride [10], sertindole [11], and grepafloxacin [12]. This unfortunate condition is both related to the physiological importance of the channel and its three-dimensional structure. The hERG channel has high ligand promiscuity, mainly due to its large hydrophobic intracellular binding pocket and its multiple states (open, inactive, and closed) [13].
The high ligand promiscuity and risk of sudden death triggered great concern for hERG liability. Assessment of chemically induced QT interval prolongation and potential proarrhythmic risk has been made mandatory in clinical trials since 2005 by the US Food and Drug Administration (FDA) [14, 15]. There are several in vitro and in vivo assays performed during preclinical studies for evaluation of hERG [14]. Among the in vitro assays, conventional patch-clamp electrophysiology remains the preferred method. This method evaluates the electric current passing through hERG channels expressed in cells, before and after test compound addition [16]. The most common cell lines include human embryonic kidney (HEK) 293 cells, Chinese hamster ovary (CHO) cells, or Xenopus laevis oocytes (XO) cells. Usually, these cells present similar outcome and discrepancies are related with poor standardization of protocols [17, 18]. In vivo electrophysiology assays are usually performed in dogs and monkeys, but other species are used as well. The ionic mechanisms of repolarization in adult rats and mice differ from those in humans. Therefore, these animals are not suited for evaluation of hERG [14]. These assays are often delayed until the late stage of preclinical development, owing to cost and resource constraints. As an alternative, transgenic zebrafish models have been validated as a high-throughput animal model [19].
Despite all the testing guidelines, hERG blockage is still missed in clinical trials and may go undiscovered for years. Recent studies have shown that current in vitro and in vivo assays are not fully predictive of potential arrhythmia risks [5, 20]. For instance, rubidium efflux has shown to be oversensitive [21] and fluorescence assays are prone to experimental artifacts [22]. In addition, in general, drugs with faster overall kinetics and drugs with higher affinity for the open state than to the inactivated state induce more QT prolongation. These characteristics of drug-hERG interaction are likely to be more arrhythmogenic but cannot be predicted by IC50 measurement alone [23]. To overcome this issue, there have been efforts to develop new in vitro assays that aim to modernize and improve the quality of early hERG evaluation [5]. The Comprehensive in vitro Proarrhythmia Assay (CiPA) considers an approach involving a multiple ion channel panel, in silico evaluation, human cardiomyocyte stem cell assays, and in human phase 1 ECGs [24]. Despite some progress, experimental testing is still expensive and time consuming, which has prompted scientists to search for more cost-effective and mechanistic paradigms to assess drug liability. As a practical solution, computational approaches have earned recognition to manage chemical-biological data and evaluate chemicals not yet tested in any experimental protocol [25].
3.2 Computational Approaches
The recent increase of hERG blockage data, mostly produced by automated hERG assays [26], has been contributing to a rapid increase in the number of compounds tested, helping populate bioactivity chemical databases such as ChEMBL [27]. These large datasets promote great possibilities for computational studies, such as structure-activity relationship (SAR) analysis, quantitative structure-activity relationship (QSAR) modeling, and pharmacophore modeling [28]. The publicly available hERG data jumped from 15 molecules [29], when the first in silico model was developed (2002), to 6690, as used by the most recent study [30]. As of date, a full crystal structure of hERG is not available yet, but structural details for hERG and molecular docking have been performed by homology modeling [31].
3.3 Ligand-Based Approaches
Most of the ligand-based approaches consist of QSAR models, but several pharmacophore models have been proposed as well. QSAR modeling is an important approach used to design novel bioactive compounds or to evaluate chemical safety. This approach employs statistical or machine learning techniques to establish predictive correlations between intrinsic chemical properties (chemical descriptors) and measured bioactivity or biological properties, such as toxicity, and the resulting models are used to predict the respective target properties of novel or untested compounds [32, 33]. Several QSAR models to predict hERG blockage have been published over the years. A comprehensive analysis of all the publications up to 2013 of these models has been made by our group and published elsewhere [34].
As of 2014, a critical analysis reveals that the vast majority of the published QSAR models do not comply with the standard validation procedures and the different statistical criteria described in the best practices of QSAR modeling [35, 36]. Most of those models are indeed not compliant with the Organization for Economic Co-operation and Development (OECD) guidance on QSAR model development and validation [37]. More specifically, the primary drawbacks of the majority of published QSAR studies are as follows: (i) most models do not have proof of passing the Y-randomization test [38–71]; (ii) no proof of applicability domain (AD) estimation is provided [38–63, 71–75]; and (iii) model predictivity is not acceptable [76–78]. As a consequence, despite a significant number of QSAR models for hERG blockage being available in the literature, only very few models could actually be employed to predict hERG blockage [77, 79–81]. Also, most of the models and associated datasets used to build them are not available online to the scientific community.
Here, we update the comprehensive analysis of QSAR models for predicting hERG blockage recently made by our group [34], commenting on the most interesting works published recently. The summary of these publications can be found in Table 3.1. All these studies used collated data on inhibition of hERG K+ channels on various cells evaluated by using the patch-clamp technique.
Table 3.1 QSAR studies for predicting hERG blockage, published during the period 2014-2016
| Number of compounds | Dataset reference/database | Descriptors | Statistical/ML approach | Performance | AD | Y-rand | References |
| 236 | Various | Structural, atomic, conformational | DT | Se = 0.88, 0.75 | Yes | No | [82] |
| Sp = 0.92, 0.94 | |||||||
| 1374 | PLR | Se = 0.90, 0.84, 0.86 | |||||
| Sp = 0.83, 0.84, 0.67 | |||||||
| 2644 | [83] | Global, ECFP | LCB | Acc = 0.91 | No | No | [84] |
| Se = 0.90 | |||||||
| Sp = 0.92 | |||||||
| 4899 | ChEMBL, WOMBAT-PK | MACCS, FeatMorgan, Pharmacophore, PubChem | SVM, RF, Tree bagging, GBM | BAC = 0.83−0.93 | Yes | Yes | [34] |
| 5,984 | ChEMBL | Morgan, CDK | SVM | BACbinary ≈ 0.8 | Yes | Yes | [85] |
| Accmulticlass ≈ 0.7 | |||||||
| 239 | Tox-database | Experimental data, PADEL | ANN | R2 = 0.68 | No | No | [86] |
| RMSE = 0.78 | |||||||
| NRMSE = 11.25% | |||||||
| 400 | [71] | CORAL | Monte Carlo | Yes | No | [87] | |
| 172 | OCHEM, Fenitchel | PADEL | kNN | Se = 0.63 | No | No | [88] |
| Sp = 0.54 | |||||||
| 6690 | ChEMBL | Physicochemical, topological | GBM | Acc = 0.72−0.78 | No | No | [30] |
| Se = 0.75−0.87 | |||||||
| Sp = 0.64−0.81 | |||||||
| 587 | [60] | Pharmacophore | RP | AUC = 0.76−0.83 | No | No | [89] |
| NB | Se = 0.79−0.90 | ||||||
| SVM | Sp = 0.69−0.72 |
Acc, accuracy; AD, applicability domain; ANN, artificial neural networks; BAC, balanced accuracy; CDK, chemistry development kit; DT, decision tree; ECFP, extended connectivity fingerprints; GBM, gradient-boosting method; kNN, k-nearest neighbors; LCB, Laplacian-corrected Bayesian; ML, machine learning; NB, naïve Bayes; NRMSE, normalized root-mean-square error; PLR, partial logistic regression; RF, random forest; RMSE, root-mean-square error; RP, recursive partitioning; Se, sensitivity; Sp, specificity; SVM, support vector machines; Y-rand, Y-randomization.
A critical analysis of recent models reveals that most of the models present the same failures as reported in the previous study [34]. These major drawbacks compromise the practical use of these models for reliable assessment of drug-induced QT syndrome. A few improvements are worth commenting on. It has been shown that acids and zwitterions have less affinity for hERG than bases and neutral compounds [90]. Nikolov et al. [82] showed that a higher predictivity of local QSAR model using a decision tree over a global QSAR model based on a larger diverse training set for acids and zwitterionic ampholytes, but at the expense of coverage. In a recent study [86], the authors developed QSAR models for predicting the blockage of multiple ion channels, including hERG. These models have been implemented in a software called the Cardiac Safety Simulator (https://www.certara.com/software/pbpk-modeling-and-simulation/cardiac-safety-simulator/), along with models for slow delayed rectifying potassium current, peak sodium current, and late calcium current. Despite the innovative approaches, the authors did not follow the best practices for model development and validation [36], not curating properly the datasets and there is no proof of Y-randomization evaluation. In another recent study [30], the authors removed from the hERG dataset extracted from ChEMBL [91] the assays known not to be predictive of hERG, which is a practice that should be considered by modelers from now on, but unfortunately the authors did not make these models publicly available.
The models developed by Braga et al. [34, 85] still represent an innovative and useful tool for the scientific community. The authors developed both binary with correct classification rate (CCR) = 0.8 and multiclass models for hERG using the ChEMBL data containing 5984 compounds. Robust and externally predictive binary (CCR = 0.8) and multiclass models (accuracy = 0.7) were developed. These models have been implemented in a public web app available at http://www.labmol.com.br/predherg. This app has been updated for this chapter and the improvements are discussed in Section 3.5.1.
Several pharmacophore models have been generated over the years [38, 48, 59, 77, 81, 89, 92–98]. A pharmacophore is, according to IUPAC [99], “an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.” A pharmacophore model can be generated by superposing a set of active molecules and extracting common chemical features that are essential for their bioactivity or by probing possible interaction points between the macromolecules of the active ligands and target. Most of the pharmacophore hypotheses reported for hERG were generated using a limited number of compounds. They also were typically shown to be less predictive than QSAR models. A few groups have integrated pharmacophore models as descriptors for QSAR models [48, 77, 81, 89].
3.4 Structure-Based Approaches
Owing to the absence of hERG experimental 3D structures, homology modeling techniques have been applied to potassium channels that were used as templates. Several homology models have been proposed so far [100–104], most of them better contributing to comprehending possible channel functioning and drug binding interactions rather than being used as a predictive tool. The structure of hERG is proposed as consisting of a tetramer containing six α-helical transmembrane segments defined as S1–S6. The voltage sensor domain (VSD) is embedded by the S1-S4 segments, which defines the open, closed, and inactivated states. These states are administered by the positively charged lysine and arginine residues in the S4 helix in response to changes in the membrane potential. The pore domain is formed by the S5 and S6 segments, which allows potassium ions to cross the membrane. A selectivity filter loop is present in these segments along with a S5-P linker. Both N- and C-terminal domains are located on the intracellular side of the membrane [1, 105].
Usually, hERG inhibitors interact with the pore module. Several homology models indicated that important amino acids include T623, S624, and V625 from the P1, and residues G648, Y652, and F656 located on the helix S6 [106–108]. A recent study indicated that there is a high-affinity aromatic binding determinant for blockers located in helix S5, F557 [31]. Common templates for hERG channels are the crystal structure of KcsA of Streptomyces lividans in the closed state [109], KvAP of Mus musculus [110], MthK of Methanothermobacter [111], and more recently the Kv1.2 of Rattusnorvegicus [112, 113]. The last three are in the open state. A structural representation of the hERG channel is presented in Figure 3.1.

Figure 3.1 Structural representation of hERG channel generated through homology modeling. This model was generated using the open conformation of the hERG channel (UniProt accession number: Q12809) and the KvAP crystal structure (PDB code: 1ORQ) of Mus musculus [110] as template. The model was generated using a similar protocol reported by Farid et al. [102]. (a) Tetrameric representation of hERG channel. (b) Dimeric representation of S5 and S6 segments. The residues usually involved in drug interaction are represented by sticks. Each black sphere represents a potassium ion. (See color plate section for the color representation of this figure.)
3.5 Applications to Predict hERG Blockage
Several tools, essentially from academic groups, are freely available to predict the hERG blockage. We can also mention commercial tools such as StarDrop - ADME QSAR models (Optibrium Ltd., http://www.optibrium.com/stardrop/), which has a hERG model that predicts pIC50 values for inhibition of hERG K+ channels expressed in mammalian cells. This model uses 158 descriptors, including a mixture of physicochemical properties and fingerprints. The model was trained using 135 compounds, with 35 compounds in the test set. The quality of the models during the external validation achieved a Q2 of 0.72 and RMSE of 0.64 log unit. An important feature of the StarDrop module is the ability to use a user's hERG data to improve the built-in model. Another commercial tool is QuikProp from Schrödinger Suite (Schrödinger, LLC, http://www.schrodinger.com/), designed to predict pIC50 values for inhibition of hERG K+ channels expressed in mammalian cells. The model was trained using 47 compounds, and the quality of the models is R2 of 0.76 and RMSE of 0.8 log unit. The AdmetSAR (http://lmmd.ecust.edu.cn:8000) is an open webserver for many endpoints and it has a binary QSAR classification model for hERG blockage, developed from an academic group [114]. The model was trained using 368 molecules including 79 strong hERG inhibitors (pIC50 > 6.0 mol/L) and 289 weak hERG inhibitors (pIC50 ≤ 6.0 mol/L). The Pred-hERG web app (http://www.labmol.com.br/predherg) is an app developed by the LabMol - Laboratory of Molecular Modeling and Drug Design of the Federal University of Goias, Brazil [34, 85]. This app is based on statistically significant and externally predictive QSAR models of hERG blockage. The models were built using the largest publicly available dataset of structurally diverse compounds including a variety of drug classes (see Section 3.6 for more information).
3.5.1 Pred-hERG Web App
The Pred-hERG is a web app available for early identification of putative hERG blockers and non-blockers in chemical libraries, based on binary and multiclass QSAR models. Recently, we released Pred-hERG 4.0, an enhanced version based on ChEMBL v.21 and other public data with 16,932 chemical records in raw data. After a careful data curation [115], 8489 compounds including 4437 non-blockers (activity ≥ 10 μM), 2753 weak/moderate blockers (1 μM ≤ activity ≤ 10 μM), and 1299 strong blockers (activity ≤ 1 μM) remained for the modeling. To the best of our knowledge, this is the largest publicly available hERG dataset [85, 116]. We succeeded in developing a robust and externally predictive binary (CCR ≈ 0.82) and multiclass models (accuracy ≈ 0.7). These models are available at Pred-hERG web app, which is freely available to the public at http://www.labmol.com.br/predherg. Prediction time for a compound is just a few seconds. Three following outcomes are available for the users: prediction by binary model, prediction by multiclass model, and the probability maps of atomic contribution (Figure 3.2).

Figure 3.2 Outcome interpretation from the Pred-hERG web app. Binary prediction, multiclass prediction, and predicted probability maps (PPM) extracted from binary models using Morgan fingerprints with 2048 bits. In the PPMs, green atoms or fragments represent contribution toward blockage of hERG, while pink indicate that it contributes to decrease of hERG blockage, and gray means no contribution. Gray isolines delimit the region of split between the positive (green) and the negative (pink) contribution. (See color plate section for the color representation of this figure.)
As shown in Figure 3.2, the Pred-hERG web app provides highly accurate predictions along with predicted probability of fragment contributions represented in a figure, allowing end users to better understand the prediction and propose new compounds. For binary models, the probability of the compound to be a hERG non-blocker or blocker is reported in parenthesis (in this order). The predicted probability represents the result of a consensus prediction for 500 decision trees from a random forest model (e.g., 375 decision trees predicted the compound to be blocker, so the probability to be blocker is 75%). Multiclass models have similar outcome, but the user will receive the probability of a compound to be a non-blocker, weak/moderate blocker, or strong blocker, respectively. The association of fragment importance derived from QSAR models clearly helps in ending the stigma attached to QSAR as a “black box.”
Recently, we have shown that toxicity prediction solely based on structural alerts is unreliable in most cases and should be avoided [117]. Therefore, alerts should be used only as a hypothesis of toxicity mechanism and after validation by QSAR. For illustrating this, we selected two well-known structural alerts for hERG and compared them with predictions made by QSAR. The presence of a tertiary amine is one of the most described substructure alerts for hERG blockage [50, 118]. Previously [34], we presented 16 examples showing in detail the electronic and steric change in the environment of the tertiary amine that could transform a potent hERG blocker to a less potent blocker or even to a non-blocker compound. Figure 3.3a shows the compares theuse of a tertiary amine as a predictor of hERG blockage versus the predictive power of QSAR models. In the studied dataset, we found 5984 compounds (3436 blockers and 2548 non-blockers) with at least one tertiary amine. Employing only the alert, the probability of success in selecting a hERG blocker from this dataset is 57%. To evaluate the true predictive power of the developed QSAR models, we reanalyzed the external sets during the fivefold external cross-validation, quantifying the predictions only for the compounds having at least one tertiary amine moiety for each external fold. We found a probability of 84% of success in selecting a hERG blocker (selectivity ≈ 0.84), with a good correct classification rate (CCR ≈ 0.8) and good ability to classify non-blocker compounds as well (specificity ≈ 0.76).
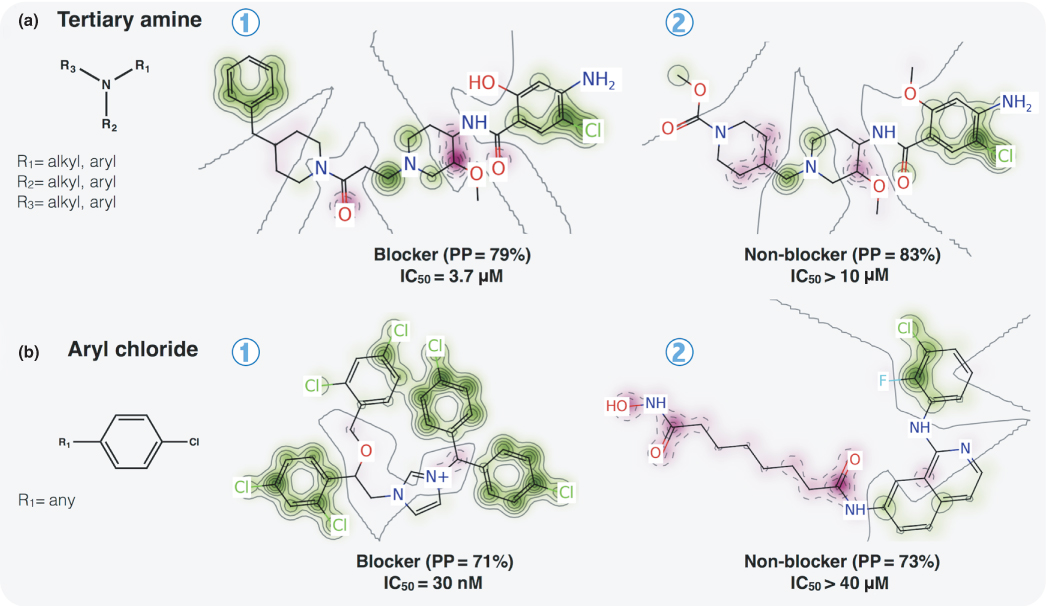
Figure 3.3 Comparison of structural alerts and the Pred-hERG QSAR models for prediction of hERG blockage. (a) Tertiary amines. (b) Aryl chloride. PP, predicted probability. (See color plate section for the color representation of this figure.)
The Pred-hERG combines quantitative toxicity prediction with a visualization output, ensuring both high predictivity and interpretability of models. In the PPMs, green atoms or fragments represent a contribution toward blockage of hERG, while pink indicate contribution to decrease of hERG blockage, and gray indicate no contribution. Gray isolines delimit the region of the split between the positive (green) and the negative (pink) contributions (see Figure 3.2).
Owing to its high predictive power associated with a visualization method that allows easy model interpretation, Pred-hERG represents an innovative tool to predict hERG blockage contributing to the design of safer compounds. Furthermore, Pred-hERG 4.0 implements the largest publicly available dataset for hERG blockage, which spans the largest chemical space in terms of available tools for the use of the scientific community.
3.6 Other Computational Approaches Related to hERG Liability
In addition to ligand and structure-based approaches to predict and/or understand characteristics that influence hERG blockage, other computational approaches have been proposed to better comprehend this phenomenon. Recently [119], a data mining approach that identifies from electronic health records (EHRs) common clinical features associated with the use of drugs that do or do not prolong the QT interval identified that the combination of two drugs not associated with QT prolongations appeared to increase the liability for long QT syndrome. EHR offer unprecedented opportunities to comprehend post-marketed drug issues [120]. As another example, Romero et al. [121] developed a computational approach to reveal the pharmacological properties of drugs that are most likely to induce long QT prolongation that would be expected to result from genetic variants.
From now on, new computational studies must address current knowledge to better predict hERG blockage. The CiPA recommends that in silico models of the human ventricular myocyte should use data obtained at physiological temperatures, but as temperature may have an effect on channel gating and drug binding rate [122], a base model dealing with temperature-dependent gating changes without drugs and a pharmacodynamics component simulating temperature-dependent drug binding kinetics should be used [123].
The role of hERG is well characterized in ventricular repolarization. However, other ion channels are also related to the cardiac electrical activity, and, therefore, evaluation of Torsades de Pointes based only on hERG appears now to be not enough to predict cardiac toxicity [124]. Few computational studies have introduced multiple ion approaches to predict cardiac toxicity [71, 86], but well-validated models are required to use these models on a large scale. We suggest that current hERG models should be used along with other ion channels models to better predict cardiac toxicity.
3.7 Final Remarks
Despite some progress made in the last decade to predict hERG blockage, we observe that these efforts still do not fully guarantee that new chemicals do not induce QT prolongation. The absence of a crystal structure of hERG associated with current flaws in experimental assays reveals that there is significant scope for development of innovative studies using modern data and considering key information gathered in recent years. Certainly, the newly proposed experimental assays for hERG will increase the quality and the correlation of these assays with human response, allowing computational modelers to generate more comprehensive and predictive models.
References
- 1 Vandenberg, J.I., Perry, M.D., Perrin, M.J. et al. (2012) hERG K+ channels: structure, function, and clinical significance. Physiol. Rev., 92, 1393–1478.
- 2 Tso, C., Currie, G.M., Gilmore, D., and Kiat, H. (2015) Electrocardiography: a technologist's guide to interpretation. J. Nucl. Med. Technol., 43, 247–252.
- 3 Hedley, P.L., Jørgensen, P., Schlamowitz, S. et al. (2009) The genetic basis of long QT and short QT syndromes: a mutation update. Hum. Mutat., 30, 1486–1511.
- 4 Nachimuthu, S., Assar, M.D., and Schussler, J.M. (2012) Drug-induced QT interval prolongation: mechanisms and clinical management. Ther. Adv. Drug. Saf., 3, 241–253.
- 5 Lester, R.M. and Olbertz, J. (2016) Early drug development: assessment of proarrhythmic risk and cardiovascular safety. Expert Rev. Clin. Pharmacol., 9, 1611–1618.
- 6 Brown, A.M. (2004) Drugs, hERG and sudden death. Cell Calcium, 35, 543–547.
- 7 Siramshetty, V.B., Nickel, J., Omieczynski, C. et al. (2016) WITHDRAWN—a resource for withdrawn and discontinued drugs. Nucleic Acids Res., 44, D1080–D1086.
- 8 Stockbridge, N., Morganroth, J., Shah, R.R., and Garnett, C. (2013) Dealing with global safety issues: was the response to QT-liability of non-cardiac drugs well coordinated? Drug Saf., 36, 167–182.
- 9 Woosley, R.L. (1996) Cardiac actions of antihistamines. Annu. Rev. Pharmacol. Toxicol., 36, 233–252.
- 10 Rampe, D., Roy, M.L., Dennis, A., and Brown, A.M. (1997) A mechanism for the proarrhythmic effects of cisapride (Propulsid): high affinity blockade of the human cardiac potassium channel HERG. FEBS Lett., 417, 28–32.
- 11 Alvarez, P.A. and Pahissa, J. (2010) QT alterations in psychopharmacology: proven candidates and suspects. Curr. Drug Saf., 5, 97–104.
- 12 Roden, D.M. (2004) Drug-induced prolongation of the QT interval. N. Engl. J. Med., 350, 1013–1022.
- 13 Mitcheson, J.S., Chen, J., Lin, M. et al. (2000) A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA, 97, 12329–12333.
- 14 FDA (2005) Guidance for industry. S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals, Rockville, MD.
- 15 FDA (2005) E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Rockville, MD.
- 16 Hamill, O.P., Marty, A., Neher, E. et al. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch., 391, 85–100.
- 17 Wiśniowska, B. and Polak, S. (2009) hERG in vitro interchange factors--development and verification. Toxicol. Mech. Methods, 19, 278–284.
- 18 Witchel, H.J., Milnes, J.T., Mitcheson, J.S., and Hancox, J.C. (2003) Troubleshooting problems with in vitro screening of drugs for QT interval prolongation using HERG K+ channels expressed in mammalian cell lines and Xenopus oocytes. J. Pharmacol. Toxicol. Methods, 48, 65–80.
- 19 Wen, D., Liu, A., Chen, F. et al. (2012) Validation of visualized transgenic zebrafish as a high throughput model to assay bradycardia related cardio toxicity risk candidates. J. Appl. Toxicol., 32, 834–842.
- 20 Takasuna, K., Asakura, K., Araki, S. et al. (2016) Comprehensive in vitro cardiac safety assessment using human stem cell technology: Overview of CSAHi HEART initiative. J. Pharmacol. Toxicol. Methods, 83, 42–54.
- 21 Chaudhary, K.W., O'Neal, J.M., Mo, Z.-L. et al. (2006) Evaluation of the rubidium efflux assay for preclinical identification of HERG blockade. Assay Drug Dev. Technol., 4, 73–82.
- 22 Polak, S., Wiśniowska, B., and Brandys, J. (2009) Collation, assessment and analysis of literature in vitro data on hERG receptor blocking potency for subsequent modeling of drugs' cardiotoxic properties. J. Appl. Toxicol., 29, 183–206.
- 23 Lee, W., Mann, S.A., Windley, M.J. et al. (2016) In silico assessment of kinetics and state dependent binding properties of drugs causing acquired LQTS. Prog. Biophys. Mol. Biol., 120, 89–99.
- 24 Crumb, W.J., Vicente, J., Johannesen, L., and Strauss, D.G. (2016) An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods, 81, 251–262.
- 25 Naven, R. and Louise-May, S. (2015) Computational toxicology: its essential role in reducing drug attrition. Hum. Exp. Toxicol., 34, 1304–1309.
- 26 Li, X., Zhang, R., Zhao, B. et al. (2016) Cardiotoxicity screening: a review of rapid-throughput in vitro approaches. Arch. Toxicol., 90, 1803–1816.
- 27 Gaulton, A., Bellis, L.J., Bento, A.P. et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res., 40, D1100–D1107.
- 28 Fourches, D. (2014) Cheminformatics: at the crossroad of eras, in Application of Computational Techniques in Pharmacy and Medicine (eds L. Gorb, V. Kuz'min, and E. Muratov), Springer Netherlands, Dordrecht, pp. 539–546.
- 29 Ekins, S., Crumb, W.J., Sarazan, R.D. et al. (2002) Three-dimensional quantitative structure-activity relationship for inhibition of human ether-a-go-go-related gene potassium channel. J. Pharmacol. Exp. Ther., 301, 427–434.
- 30 Didziapetris, R. and Lanevskij, K. (2016) Compilation and physicochemical classification analysis of a diverse hERG inhibition database. J. Comput. Aided Mol. Des., 30, 1175–1188.
- 31 Saxena, P., Zangerl-Plessl, E.-M., Linder, T. et al. (2016) New potential binding determinant for hERG channel inhibitors. Sci. Rep., 6, 24182.
- 32 Cherkasov, A., Muratov, E.N., Fourches, D. et al. (2014) QSAR modeling: where have you been? Where are you going to? J. Med. Chem., 57, 4977–5010.
- 33 Dearden, J.C. (2016) The history and development of quantitative structure–activity relationships (QSARs). Int. J. Quant. Struct. Relationships, 1, 1–44.
- 34 Braga, R.C., Alves, V.M., Silva, M.F.B. et al. (2014) Tuning HERG out: antitarget QSAR models for drug development. Curr. Top. Med. Chem., 14, 1399–1415.
- 35 Golbraikh, A. and Tropsha, A. (2002) Beware of q2!. J. Mol. Graph Model., 20, 269–276.
- 36 Tropsha, A. (2010) Best practices for QSAR model development, validation, and exploitation. Mol. Inform., 29, 476–88.
- 37 OECD. OECD principles for the validation, for regulatory purposes, of (quantitative) structure–activity relationship models 2004, pp. 1–2. http://www.oecd.org/chemicalsafety/risk-assessment/37849783.pdf (accessed July 31, 2017).
- 38 Cavalli, A., Poluzzi, E., De Ponti, F., and Recanatini, M. (2002) Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem., 45, 3844–3853.
- 39 Pearlstein, R.A., Vaz, R.J., Kang, J. et al. (2003) Characterization of HERG potassium channel inhibition using CoMSiA 3D QSAR and homology modeling approaches. Bioorg. Med. Chem. Lett., 13, 1829–1835.
- 40 Bains, W., Basman, A., and White, C. (2004) HERG binding specificity and binding site structure: evidence from a fragment-based evolutionary computing SAR study. Prog. Biophys. Mol. Biol., 86, 205–233.
- 41 Tobita, M., Nishikawa, T., and Nagashima, R. (2005) A discriminant model constructed by the support vector machine method for HERG potassium channel inhibitors. Bioorg. Med. Chem. Lett., 15, 2886–2890.
- 42 O'Brien, S.E. and de Groot, M.J. (2005) Greater than the sum of its parts: combining models for useful ADMET prediction. J. Med. Chem., 48, 1287–1291.
- 43 Cianchetta, G., Li, Y., Kang, J. et al. (2005) Predictive models for hERG potassium channel blockers. Bioorg. Med. Chem. Lett., 15, 3637–3642.
- 44 Coi, A., Massarelli, I., Murgia, L. et al. (2006) Prediction of hERG potassium channel affinity by the CODESSA approach. Bioorg. Med. Chem., 14, 3153–3159.
- 45 Ekins, S., Balakin, K.V., Savchuk, N., and Ivanenkov, Y. (2006) Insights for human ether-a-go-go-related gene potassium channel inhibition using recursive partitioning and Kohonen and Sammon mapping techniques. J. Med. Chem., 49, 5059–5071.
- 46 Gepp, M.M. and Hutter, M.C. (2006) Determination of hERG channel blockers using a decision tree. Bioorg. Med. Chem., 14, 5325–5332.
- 47 Seierstad, M. and Agrafiotis, D.K. (2006) A QSAR model of HERG binding using a large, diverse, and internally consistent training set. Chem. Biol. Drug Des., 67, 284–296.
- 48 Leong, M.K. (2007) A novel approach using pharmacophore ensemble/support vector machine (PhE/SVM) for prediction of hERG liability. Chem. Res. Toxicol., 20, 217–226.
- 49 Inanobe, A., Kamiya, N., Murakami, S. et al. (2008) In silico prediction of the chemical block of human ether-a-go-go-related gene (hERG) K+ current. J. Physiol. Sci., 58, 459–470.
- 50 Li, Q., Jørgensen, F.S., Oprea, T. et al. (2008) hERG classification model based on a combination of support vector machine method and GRIND descriptors. Mol. Pharm., 5, 117–127.
- 51 Jia, L. and Sun, H. (2008) Support vector machines classification of hERG liabilities based on atom types. Bioorg. Med. Chem., 16, 6252–6260.
- 52 Thai, K.-M. and Ecker, G.F. (2009) Similarity-based SIBAR descriptors for classification of chemically diverse hERG blockers. Mol. Divers., 13, 321–336.
- 53 Fenu, L.A., Teisman, A., De Buck, S.S. et al. (2009) Cardio-vascular safety beyond hERG: in silico modelling of a guinea pig right atrium assay. J. Comput. Aided Mol. Des., 23, 883–895.
- 54 Ermondi, G., Visentin, S., and Caron, G. (2009) GRIND-based 3D-QSAR and CoMFA to investigate topics dominated by hydrophobic interactions: the case of hERG K+ channel blockers. Eur. J. Med. Chem., 44, 1926–1932.
- 55 Su, B.-H., Shen, M., Esposito, E.X. et al. (2010) In silico binary classification QSAR models based on 4D-fingerprints and MOE descriptors for prediction of hERG blockage. J. Chem. Inf. Model., 50, 1304–1318.
- 56 Kim, J.-H., Chae, C.-H., Kang, S.-M. et al. (2011) The predictive QSAR model for hERG inhibitors using Bayesian and random forest classification method. Bull Korean Chem Soc, 32, 1237–1240.
- 57 Su, B.-H., Tu, Y., Esposito, E.X., and Tseng, Y.J. (2012) Predictive toxicology modeling: protocols for exploring hERG classification and Tetrahymena pyriformis end point predictions. J. Chem. Inf. Model., 52, 1660–1673.
- 58 Broccatelli, F., Mannhold, R., Moriconi, A. et al. (2012) QSAR modeling and data mining link Torsades de Pointes risk to the interplay of extent of metabolism, active transport, and hERG liability. Mol. Pharm., 9, 2290–2301.
- 59 Tan, Y., Chen, Y., You, Q. et al. (2012) Predicting the potency of hERG K+ channel inhibition by combining 3D-QSAR pharmacophore and 2D-QSAR models. J. Mol. Model., 18, 1023–1036.
- 60 Wang, S., Li, Y., Wang, J. et al. (2012) ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharm., 9, 996–1010.
- 61 Wang, Z., Mussa, H.Y., Lowe, R. et al. (2012) Probability based hERG blocker classifiers. Mol. Inform., 31, 679–685.
- 62 Coi, A. and Bianucci, A.M. (2013) Combining structure- and ligand-based approaches for studies of interactions between different conformations of the hERG K(+) channel pore and known ligands. J. Mol. Graph. Model., 46, 93–104.
- 63 Polak, S., Wiśniowska, B., Glinka, A. et al. (2013) Slow delayed rectifying potassium current (IKs) - analysis of the in vitro inhibition data and predictive model development. J. Appl. Toxicol., 33, 723–739.
- 64 Dubus, E., Ijjaali, I., Petitet, F., and Michel, A. (2006) In silico classification of HERG channel blockers: a knowledge-based strategy. ChemMedChem, 1, 622–630.
- 65 Obrezanova, O., Csanyi, G., Gola, J.M.R., and Segall, M.D. (2007) Gaussian processes: a method for automatic QSAR modeling of ADME properties. J. Chem. Inf. Model., 47, 1847–1857.
- 66 Thai, K.-M. and Ecker, G.F. (2008) A binary QSAR model for classification of hERG potassium channel blockers. Bioorg. Med. Chem., 16, 4107–4119.
- 67 Chekmarev, D.S., Kholodovych, V., Balakin, K.V. et al. (2008) Shape signatures: new descriptors for predicting cardiotoxicity in silico. Chem. Res. Toxicol., 21, 1304–1314.
- 68 Hansen, K., Rathke, F., Schroeter, T. et al. (2009) Bias-correction of regression models: a case study on hERG inhibition. J. Chem. Inf. Model., 49, 1486–1496.
- 69 Nisius, B. and Göller, A.H. (2009) Similarity-based classifier using topomers to provide a knowledge base for hERG channel inhibition. J. Chem. Inf. Model., 49, 247–256.
- 70 Robinson, R.L.M., Glen, R.C., and Mitchell, J.B.O. (2011) Development and comparison of hERG blocker classifiers: assessment on different datasets yields markedly different results. Mol. Inform., 30, 443–458.
- 71 Obiol-Pardo, C., Gomis-Tena, J., Sanz, F. et al. (2011) A multiscale simulation system for the prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model., 51, 483–492.
- 72 Yap, C.W., Cai, C.Z., Xue, Y., and Chen, Y.Z. (2004) Prediction of torsade-causing potential of drugs by support vector machine approach. Toxicol. Sci., 79, 170–177.
- 73 Song, M. and Clark, M. (2006) Development and evaluation of an in silico model for hERG binding. J. Chem. Inf. Model., 46, 392–400.
- 74 Gunturi, S.B., Archana, K., Khandelwal, A., and Narayanan, R. (2008) Prediction of hERG potassium channel blockade using kNN-QSAR and local lazy regression methods. QSAR Comb. Sci., 27, 1305–1317.
- 75 Czodrowski, P. (2013) hERG me out. J. Chem. Inf. Model., 53, 2240–2251.
- 76 Gavaghan, C.L., Arnby, C.H., Blomberg, N. et al. (2007) Development, interpretation and temporal evaluation of a global QSAR of hERG electrophysiology screening data. J. Comput. Aided Mol. Des., 21, 189–206.
- 77 Du-Cuny, L., Chen, L., and Zhang, S. (2011) A critical assessment of combined ligand- and structure-based approaches to HERG channel blocker modeling. J. Chem. Inf. Model., 51, 2948–2960.
- 78 Kar, S. and Roy, K. (2012) Prediction of hERG potassium channel blocking actions using combination of classification and regression based models: a mixed descriptors approach. Mol. Inform., 31, 879–894.
- 79 Sinha, N. and Sen, S. (2011) Predicting hERG activities of compounds from their 3D structures: development and evaluation of a global descriptors based QSAR model. Eur. J. Med. Chem., 46, 618–630.
- 80 Pourbasheer, E., Beheshti, A., Khajehsharifi, H. et al. (2013) QSAR study on hERG inhibitory effect of kappa opioid receptor antagonists by linear and non-linear methods. Med. Chem. Res., 22, 4047–4058.
- 81 Moorthy, N.S.H.N., Ramos, M.J., and Fernandes, P.A. (2013) QSAR and pharmacophore analysis of a series of piperidinyl urea derivatives as HERG blockers and H3 antagonists. Curr. Drug Discov. Technol., 10, 47–58.
- 82 Nikolov, N.G., Dybdahl, M., Jónsdóttir, S.Ó., and Wedebye, E.B. (2014) hERG blocking potential of acids and zwitterions characterized by three thresholds for acidity, size and reactivity. Bioorg. Med. Chem., 22, 6004–6013.
- 83 Doddareddy, M.R., Klaasse, E.C., Shagufta et al. (2010) Prospective validation of a comprehensive in silico hERG model and its applications to commercial compound and drug databases. ChemMedChem, 5, 716–729.
- 84 Liu, L., Lu, J., Lu, Y. et al. (2014) Novel Bayesian classification models for predicting compounds blocking hERG potassium channels. Acta Pharmacol. Sin., 35, 1093–1102.
- 85 Braga, R.C., Alves, V.M., Silva, M.F.B. et al. (2015) Pred-hERG: a novel web-accessible computational tool for predicting cardiac toxicity. Mol. Inform., 34, 698–701.
- 86 Wiśniowska, B., Mendyk, A., Szlęk, J. et al. (2015) Enhanced QSAR models for drug-triggered inhibition of the main cardiac ion currents. J. Appl. Toxicol., 35, 1030–1039.
- 87 Gobbi, M., Beeg, M., Toropova, M.A. et al. (2016) Monte Carlo method for predicting of cardiac toxicity: hERG blocker compounds. Toxicol. Lett., 250–251, 42–46.
- 88 Chavan, S., Abdelaziz, A., Wiklander, J.G., and Nicholls, I.A. (2016) A k-nearest neighbor classification of hERG K+ channel blockers. J. Comput. Aided Mol. Des., 30, 229–236.
- 89 Wang, S., Sun, H., Liu, H. et al. (2016) ADMET evaluation in Drug discovery. 16. Predicting hERG blockers by combining multiple pharmacophores and machine learning approaches. Mol. Pharm., 13, 2855–2866.
- 90 Waring, M.J. and Johnstone, C. (2007) A quantitative assessment of hERG liability as a function of lipophilicity. Bioorg. Med. Chem. Lett., 17, 1759–1764.
- 91 Willighagen, E.L., Waagmeester, A., Spjuth, O. et al. (2013) The ChEMBL database as linked open data. J Cheminform, 5, 23.
- 92 Aronov, A.M. (2008) Tuning out of hERG. Curr. Opin. Drug Discov. Dev., 11, 128–40.
- 93 Aronov, A.M. (2006) Common pharmacophores for uncharged human ether-a-go-go-related gene (hERG) blockers. J. Med. Chem., 49, 6917–6921.
- 94 Du, L.-P., Tsai, K.-C., Li, M.-Y. et al. (2004) The pharmacophore hypotheses of I(Kr) potassium channel blockers: novel class III antiarrhythmic agents. Bioorg Med Chem Lett, 14, 4771–4777.
- 95 Durdagi, S., Duff, H.J., and Noskov, S.Y. (2011) Combined receptor and ligand-based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J. Chem. Inf. Model., 51, 463–474.
- 96 Kratz, J.M., Schuster, D., Edtbauer, M. et al. (2014) Experimentally validated hERG pharmacophore models as cardiotoxicity prediction tools. J. Chem. Inf. Model., 54, 2887–2901.
- 97 Matyus, P., Borosy, A.P., Varro, A. et al. (1998) Development of pharmacophores for inhibitors of the rapid component of the cardiac delayed rectifier potassium current. Int. J. Quantum Chem., 69, 21–30.
- 98 Peukert, S., Brendel, J., Pirard, B. et al. (2004) Pharmacophore-based search, synthesis, and biological evaluation of anthranilic amides as novel blockers of the Kv1.5 channel. Bioorg. Med. Chem. Lett., 14, 2823–2827.
- 99 Wermuth, C.G.C., Ganellin, C.C.R., Lindberg, P., and Mitscher, L. (1998) Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998). Pure Appl. Chem., 70, 1129–1143.
- 100 Schmidtke, P., Ciantar, M., Theret, I., and Ducrot, P. (2014) Dynamics of hERG closure allow novel insights into hERG blocking by small molecules. J. Chem. Inf. Model., 54, 2320–2333.
- 101 Durdagi, S., Deshpande, S., Duff, H.J., and Noskov, S.Y. (2012) Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J. Chem. Inf. Model., 52, 2760–2774.
- 102 Farid, R., Day, T., Friesner, R.A., and Pearlstein, R.A. (2006) New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem., 14, 3160–3173.
- 103 Masetti, M., Cavalli, A., and Recanatini, M. (2008) Modeling the hERG potassium channel in a phospholipid bilayer: molecular dynamics and drug docking studies. J Comput Chem, 29, 795–808.
- 104 Stary, A., Wacker, S.J., Boukharta, L. et al. (2010) Toward a consensus model of the hERG potassium channel. ChemMedChem, 5, 455–467.
- 105 Durdagi, S., Subbotina, J., Lees-Miller, J. et al. (2010) Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr. Med. Chem., 17, 3514–3532.
- 106 Sanguinetti, M.C., Chen, J., Fernandez, D. et al. (2005) Physicochemical basis for binding and voltage-dependent block of hERG channels by structurally diverse drugs. Novartis Found Symp., 266, 159–166.
- 107 Kamiya, K., Niwa, R., Morishima, M. et al. (2008) Molecular determinants of hERG channel block by terfenadine and cisapride. J. Pharmacol. Sci., 108, 301–307.
- 108 Lees-Miller, J.P., Duan, Y., Teng, G.Q., and Duff, H.J. (2000) Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol., 57, 367–374.
- 109 Doyle, D.A., Morais Cabral, J., Pfuetzner, R.A. et al. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science, 280, 69–77.
- 110 Jiang, Y., Lee, A., Chen, J. et al. (2003) X-ray structure of a voltage-dependent K+ channel. Nature, 423, 33–41.
- 111 Jiang, Y., Lee, A., Chen, J. et al. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature, 417, 515–522.
- 112 Long, S.B., Tao, X., Campbell, E.B., and MacKinnon, R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature, 450, 376–382.
- 113 Long, S.B., Campbell, E.B., and Mackinnon, R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science, 309, 897–903.
- 114 Cheng, F., Li, W., Zhou, Y. et al. (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model., 52, 3099–3105.
- 115 Fourches, D., Muratov, E., and Tropsha, A. (2010) Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model., 50, 1189–1204.
- 116 Alves, V.M., Braga, R.C., Silva, M.F.B., Muratov, E., Fourches, D., Tropsha, A. et al. (2014) Pred-hERG: A novel web-accessible computational tool for predicting cardiac toxicity of drug candidates. 248th Am. Chem. Soc. Natl. Meet., vol. Abstracts, San Francisco, CA.
- 117 Alves, V.M., Muratov, E.N., Capuzzi, S.J. et al. (2016) Alarms about structural alerts. Green Chem, 18, 4348–4360.
- 118 Springer, C. and Sokolnicki, K.L. (2013) A fingerprint pair analysis of hERG inhibition data. Chem. Cent. J., 7, 167.
- 119 Roden, D.M., Mosley, J.D., and Denny, J.C. (2016) Finding a needle in a QT interval big data haystack. J. Am. Coll. Cardiol., 68, 1765–1768.
- 120 Peissig, P.L., Santos Costa, V., Caldwell, M.D. et al. (2014) Relational machine learning for electronic health record-driven phenotyping. J. Biomed. Inform., 52, 260–270.
- 121 Romero, L., Trenor, B., Yang, P.-C. et al. (2015) In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J. Mol. Cell. Cardiol., 87, 271–282.
- 122 Windley MJ, Mann SA, Vandenberg JI, Hill A. Temperature effects on kinetics of Kv11.1 drug block have important consequences for in silico proarrhythmic risk prediction. Mol. Pharmacol. 2016. DOI: 10.1124/mol.115.103127.
- 123 Li, Z., Dutta, S., Sheng, J. et al. (2016) A temperature-dependent in silico model of the human ether-à-go-go-related (hERG) gene channel. J. Pharmacol. Toxicol. Methods, 81, 233–239.
- 124 Kramer, J., Obejero-Paz, C.A., Myatt, G. et al. (2013) MICE Models: superior to the HERG model in predicting Torsade de Pointes. Sci. Rep., 3, 2100.