
Chapter 4
Computational Toxicology for Traditional Chinese Medicine
Ni Ai and Xiaohui Fan
Pharmaceutical Informatics Institute, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang, PR China
Chapter Menu
4.1 Background, Current Status, and Challenges
In the year 2015, Dr Youyou Tu won the Nobel Prize in Physiology or Medicine for her discovery of artemisinin that has saved millions lives of those suffering malaria around the world. Despite numerous debates on the relationship between artemisinin and traditional Chinese medicine (TCM), there is no doubt that inspiration from an ancient TCM medical book documenting the application of Artemisia annua L., the original herb of artemisinin, for the treatment of malaria plays a critical role in the identification of this blockbuster drug. This new triumph reawakened global interest in TCM again, as a valuable resource for novel structural scaffold identification and complementary and alternative pharmacotherapy for various common and rare human illnesses. The recent 2015 statistics from the Ministry of Industry and Information Technology of China report that the combined sales of TCM patent prescriptions and prepared decoctions have approached approximately one-third (29.26%, more than US$ 111 billion) of the annual revenue of the national pharmaceutical industry. These data reflect the expected outcome from heavy investment in the TCM sector from the Chinese government and favored use of traditional medicines through the state medical insurance program. From a global pharmaceutical trade point of view, the picture is not so promising in that the combined export of TCM products excluding raw herbs from China only reached US$ 487 million in revenue for the year of 2015 and this was mostly concentrated in the Asia Pacific countries, suggesting that the appeal of TCM remedies to Western patients remains unclear. Across the globe, after 2000 years of clinical practice and decades of scientific studies, researchers, pharmaceutical companies, and policymakers are keener than ever to uncover the unknown scientific merits of a TCM and to then take it into modern healthcare in the twenty-first century. To do so, it is important to incorporate the traditional knowledge of TCM which has accumulated over centuries with current understanding of pharmacotherapy and ensure it meets the mainstream standards on safety and efficacy.
Compared to pharmacological studies, relatively few reports focus on toxicological consequences of TCMs. Because of their natural origin, people often think TCMs are toxicity free. Before the 1980s, the toxicology-related information was incomplete for many TCM patent prescriptions on the market and the application of these medicines caused serious uncertainty in the clinic. With increasing use of TCM, the number of adverse drug reaction (ADR) reports to the state ADR monitoring center has been climbing annually. There are a number of TCM toxicity incidents which have occurred in the past two decades, including nephrotoxicity of TCM with aristolochic acids [1], interstitial pneumonia caused by Xiao Chai Hu Decoction [2], and fatal allergic reactions to TCM injections [3]. The toxicity and ADRs of TCM impact a great number of patients and have even led to death, drawing considerable attention about TCM safety in China and around the world. Safety concerns with regard to TCM is now one of the major hurdles limiting exports of TCM patent prescriptions from China. Several TCMs with cinnabar, arsenic sulfide, aconite, and aristolochic acids have been put on the blacklist of items forbidden for import into the United States, European Union, and other countries. This increasing anxiety regarding TCM safety is due to limited information available on toxicological effects and proper clinical use. In 2013, the Medicines and Healthcare Products Regulatory Agency of United Kingdom announced a warning that the aconitine composition in Zhengtian pills may cause cardio- and nervous-system toxicity. This is not surprising as Aconite is marked as a toxic TCM that should be used with caution. However, recently some traditionally nontoxic TCMs such as Polygonum multiflorum have been associated with hepatotoxicity due to improper clinical usage [4–6]. Thus comprehensive and detailed toxicological research into TCMs is urgently needed to recognize potential toxic effects, identify toxic substances, and uncover related mechanisms, which will provide critical knowledge that will guarantee safe use of TCMs in the clinic. Furthermore, this information will play an essential role in the modernization and internationalization of TCM, which now finds a place in the strategic planning of the Chinese government.
Biochemical and histopathological tests have produced valuable phenotypic toxicity data for a few TCMs but efforts are far from complete considering there are more than 600 TCMs listed in the Chinese Pharmacopeia (2015 edition). In addition, our understanding of their toxicity is significantly lacking at the cellular and molecular level, which hampers the identification of toxic substances and elucidation of potential mechanisms involved. TCMs are different from chemical drugs in many ways. A single TCM usually is composed of hundreds of chemical substances. The complexity of the system exponentially increases when multiple TCMs are combined together to form the patent prescription under investigation. It is a challenging task to isolate, extract, and identify individual chemicals in the TCM, and then test each one for safety is practically impossible. Computational toxicology is a feasible solution in the prioritization of TCM compounds for experimental evaluation with high risk. On the basis of experimental data and chemical structures, a set of predictive computational models can be generated for various toxic endpoints, which have been extensively applied in the risk assessment of new chemical entities at various stages of drug discovery and development of time- and cost-efficient methods that will effectively increase the success rate. With the advent of state-of-the-art analytical instruments and following decades of research, structural data is now available for more than 10,000 compounds originated from over 600 TCMs in the Chinese Pharmacopeia (2015 edition) [7]. However, only a limited number of those compounds were tested for toxicity. In general, construction of these in silico models for toxicity prediction needs the integration of biological and chemical information sources of these compounds; however, data collection of TCM molecules is rather poor in terms of their quantity and quality when compared to similar information on synthetic chemicals. Taking advantage of a wide battery of previously established predictive models [8], it is possible to rebuild them using the latest algorithm or directly apply these to assess the potential activities of TCM molecules on related toxic endpoints. According to the availability of toxic mechanisms, these computational models can be divided into two categories: quantitative structure-toxicity relationship (QSAR) models and toxic-target-based models.
Computational toxicological studies on TCMs are sparsely reported. Most of these works focus on prediction of a TCM's organ toxicity including hepatoxicity, cardiotoxicity, and nephrotoxicity. Various computational algorithms are utilized to construct QSAR models for screening potential toxic compounds. Drug-induced liver injury is one of the major reasons attributed to drug candidate failure during development and even for drug withdrawal from the market after approval [9, 10]. As mentioned previously, TCM-caused liver injury have been reported in the clinic and has led to serious concern on the medication safety of TCMs. It is of urgent importance to evaluate the hepatotoxic potential of chemical substances in TCMs. Huang et al. [11] extracted the details of drugs found in the Liver Toxicity Knowledge Base with known liver injury information to establish the predictive model for hepatotoxic compounds in Chinese herbal medicine (CHM) using the random forest (RF) method. Their internal cross validation and external validation both yielded a high accuracy of 79% and 87%, respectively. More than 6000 molecules in the TCM Database@Taiwan are marked by their model with liver damaging potential. Literature mining confirms predicted liver toxicity of the top 100 molecules, which demonstrates the validity of this model. Similarly, Ye et al. [12] employed three tree-based models to predict hepatotoxic compounds in TCM. This was different from the previous study as their training set included both synthetic drugs and natural compounds from TCM. In this study, boosting trees showed good overall accuracy (82%) and better specificity compared with the other two tree algorithms (the decision tree and RF). In addition, the mixture of training chemicals is proven to yield a positive effect on prediction of the external validation set. From a novel perspective of traditional TCM theory, Liu et al. [13] addressed the problem of TCM hepatoxicity prediction holistically rather on individual compounds. Their survey of known clinical and animal studies first selected 107 hepatotoxic and 431 non-hepatotoxic CHMs using a set of defined inclusion and exclusion criteria. A logistic regression model was then trained by these CHMs and 24 independent variables that described different properties and flavors of TCM in the traditional theory, such as warm, cold, and astringent, among others. Their analysis revealed statistically significant relationships between hepatotoxic CHM with certain ancient descriptions on properties and flavors, which could be a promising tool for liver toxicity evaluation of new drugs from CHMs.
The kidney plays a key role in drug metabolism and excretion and several TCMs are linked with nephrotoxicity. To prevent toxic effects of TCM on this organ, Wang et al. [14] constructed a QSAR model by the k-nearest neighbor (kNN) and support vector machine (SVM) using a public drug side-effect database (SIDER). Both models demonstrated comparable predictivity on internal cross validation but kNN showed better performance on TCM compounds in the external set. In their study, of Zhang et al. [15] performed keyword-based searches on DrugBank, TOXNET, and PubMed to prepare a training set of nephrotoxic compounds for the construction of a nephrotoxicity model. Using SVM, the classification model for nephrotoxicity yielded reasonably good predictive capacity. Interestingly, the author has also established another classification model for renal tubular injury, one of three major pathological mechanisms for nephrotoxicity. The TCM compounds predicted with nephrotoxicity from the first model would then be subject to a secondary mechanism-oriented classification model. Out of the 10 compounds output from two-step screening, the literature supported 6 compounds that can cause tubular necrosis with various experimental or clinical evidence.
There are an increasing number of reports on cardiotoxicity of TCM, mostly focusing on known toxic TCMs based on traditional theory. The inhibition of the hERG potassium channel has been associated with cardiotoxicity of several drugs such as terfenadine. Lei et al. [16] built a computational model to predict hERG inhibition for TCM compounds. Two feature elimination algorithms (RF and SVM) were applied and model construction was performed by four separate methods, boosting tree, SVM, discriminate analysis, and RF. Their results indicated that RF yielded the QSAR model with best performance (82.5% accuracy). Screening on more than 1,600 compounds from 60 toxic TCMs suggested 72 compounds that can potentially inhibit this ion channel. This in silico hERG inhibition model is a useful tool for early identification of cardiotoxicity in TCM drug discovery. Zhang et al. [17] reported the application of an SVM model on CNS toxicity prediction of TCM. Using a training set with more than 500 chemicals, an SVM classifier with reasonable predictive ability was built for identifying CNS toxic compounds over nontoxic ones. Among 13 compounds predicted with neurotoxicity in Radix Sophorae Subprostratae, the literature verified toxic activity of four TCM compounds. For computational evaluation of acute toxicity of TCM, there is only one report available that a QSAR model was constructed and applied to over 1,600 compounds from 60 toxic TCMs. In this study, 89.7%, 10.2%, and 0.1% of TCM compounds were predicted to exhibit acute/low, medium, and high toxicity, respectively, which provides important data for guiding clinical usage of these TCM.
In clinical practice, TCM is often prescribed together with chemical drugs. Since pharmacokinetics and pharmacodynamics processes are largely unknown for most TCMs, little information is currently available on TCM–drug interactions (TDI). However, the potential presence of these interactions with high risk should not be neglected because this may lead to unexpected adverse reactions and loss of efficacy of concomitantly used chemical drugs. Our recent review on in silico models for drug–drug interactions [18] has provided an in-depth summary on the latest development on this topic, in which a general workflow is also proposed to investigate TDI. Taking Salvia miltiorrhiza as an example of a TCM, we focused on the clinically reported TDI of this commonly used medicinal herb with warfarin, the anticoagulant for treating cardiovascular diseases. Since warfarin is highly human serum albumin (HSA)-bound in the plasma, the interaction of S. miltiorrhiza with this drug may occur at this target, which could increase free drug concentration in vivo and lead to bleeding issues observed clinically. A molecular docking study of over 70 compounds in S. miltiorrhiza identified 6 compounds that specifically interact with the warfarin-binding site of HSA and fluorometric experiments later confirmed our predictions, which sheds some light on the possible mechanism of HSA-mediated S. miltiorrhiza-warfarin interactions [19]. Similarly, docking techniques have been used to study TCM compounds with drug-metabolizing enzymes CYP2D6 [20], CYP2C19 [21], and other related proteins, such as PXR [22], which provide target specific information on TDI. QSAR models have also been constructed to understand the interactions between TCM and UGT1A4 [23] and the drug transporter OATP1B1 [24]. Results from these studies indicate that special attention may be needed when selecting TCM co-administered with drugs for therapeutic applications.
As reviewed above, most of the computational toxicological studies focus on the prediction of organ toxicities of TCM compounds, mainly through machine learning methods. In addition, there have been several attempts in the evaluation of TDI due to the simultaneous binding/inhibition on important proteins participating in drug metabolism and excretion using the molecular docking technique. It is noteworthy that these in silico models usually predict toxicity potential of individual compounds in TCMs. However, TCMs or modern TCM preparations, which are complex systems with more than a hundred substances, are usually used as a whole and the relative contents of these compounds may be critical to their efficacy and toxicity, which is a difficult problem that needs to be handled by the aforementioned computational models. In addition, compounds in TCMs are secondary metabolites of the plants or animals and often share the same scaffold with diverse substitutions. The occurrence of activity cliffs is very likely to happen on these structural analogs and this needs to be kept in mind to decrease prediction errors. Oral dosing is the conventional administration route for TCMs; extensive in vivo metabolism may occur as these metabolites could be potentially active or toxic ingredients of the TCMs. Previous studies normally overlooked this important group of substances, thus a complete picture of the toxicity profile of a TCM could not be fully derived. Owing to sparse and limited experimental information on TCM compounds, most of the current computational models are trained with synthetic compounds, which could be another potentially problematic issue in these studies. Differences of their chemical space between these two sets of compounds have been reported and this raises concern regarding the applicability of these models to TCM. More efforts would be required in evaluating the impact of this problem and it might be better to construct the TCM-specific predictive models when sufficient data are available.
Computational toxicological techniques have been extensively applied in various stages of discovery and development of synthetic drugs. Owing to their effect in decreasing the time and cost related to drug toxicity, these models are now recognized as key components of the whole pipeline. Relatively speaking, TCM computational toxicology is in its early phrase and more work is warranted to demonstrate its full potential in TCM-related studies, such as deciphering the toxic mechanisms and research and development of modern TCM patent prescriptions.
4.2 Case Study: Large-Scale Prediction on Involvement of Organic Anion Transporter 1 in Traditional Chinese Medicine-Drug Interactions
In the following section, we present a case study on the application of computational techniques to evaluate the potential risk of transporter-mediated drug–TCM interactions. The organic anion transporter 1 (OAT1)-mediated drug–drug interactions have received considerable interest for their potential influence on drug efficacy and toxicity. Recently, several TCMs have been shown to modulate OAT1-transporting activity, which has raised concern about the TCM–drug interactions that may occur. However, information about OAT1-mediated TCM–drug interactions are extremely limited and large-scale evaluation has not been performed for the majority of the TCMs. In this work, we have elucidated important structural features of inhibitors for OAT1 via pharmacophore modeling based on available in vitro data. The reliability of the model was validated by an external dataset and seven out of eight known inhibitor drugs in the test set were successfully mapped to the current OAT1 inhibitor pharmacophore. All the medicinal TCMs reported in the Chinese Pharmacopoeia were then subject to screening by the validated model. The screening results revealed 144 potential OAT1 inhibitors and 11 TCMs out of 611 TCMs that could likely interfere with the ability of OAT1 to transport its substrates. Literature curation confirmed the OAT1 inhibitory activities for five of these identified TCMs, suggestive of TCM–drug interactions mediated by OAT1. Further study would be required to exploit the clinical relevance of OAT1-mediated TCM–drug interactions.
4.2.1 Introduction to OAT1 and TCM
Recently, the United States Food and Drug Administration (FDA) and European Medicines Agency issued guidelines for the investigational conditions that are required prior to approval and both agencies involved the potential drug–drug interactions (DDIs) mediated by seven identified transporters, including organic anion transporter 1 and 3 (OAT1 and 3), organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1 and 1B3), organic cation transporter 2 (OCT2), multidrug resistance transporter 1 (MDR1), and the breast cancer resistance protein (BCRP) [1, 25]. Clearly, regulatory authorities have recognized the potential clinical significance of transporter-mediated DDIs and their impact on drug safety based on results from many preclinical and clinical studies, as well as post-marketing reports [26–29]. By modulating transporter activities, these DDIs may influence the pharmacokinetics of the concomitantly used drugs and further lead to adverse effects by affecting absorption, distribution, metabolism, and excretion (ADME) of drugs. To predict transporter-mediated DDIs, many studies have been conducted to characterize these transporters, identify their substrates or inhibitors, and elucidate drug specificity [30–32]. Obviously, drugs that are currently used in the clinic have been the major focus of these studies.
TCM has been practiced in Eastern Asian countries for centuries and it still significantly contributes to the current healthcare system in China with great efficacy in the treatment of various diseases, such as infections, cardiovascular diseases, and diabetes. [33–35]. With their increasing use as health supplements and complementary/alternative medicines in Western countries, we are currently observing more concomitant occurrence of TCMs and drugs in a clinical setting [36, 37]; therefore, more attention is required regarding the potential adverse effects related to the combination of TCMs and drugs. Similar to DDIs, it is possible that transporter-mediated TCM–drug interactions also occur and lead to altered blood drug concentration and plasma half-life, therefore influencing the therapeutic effects of drugs or causing adverse events [38–40]. For drugs with a narrow therapeutic index, such as digoxin, aminophylline, and cyclosporin A, it is especially important to monitor for adverse drug reactions when used in combination with TCM [41–43]. However, it is difficult to evaluate TCM–drug interactions as only limited information is available for clinical practitioners and researchers to make decisions regarding the combinational usage of TCM and drugs. Moreover, the potential for TCM–drug interactions poses a threat to the general public owing to the false impression of safety induced by the natural origin of TCM. Therefore, it is critical to evaluate transporter-mediated TCM–drug interactions, including molecular mechanisms and the possible individual molecule perpetrators in TCMs, in order to establish a reliable system to evaluate the safety of TCM–drug usage.
As one of the major pathways for drug elimination, active secretion in the proximal tubules of the kidney, particularly transporter-mediated active secretion, has been investigated for its involvement in DDI [44, 45]. Significant attention has focused on OAT1 [45, 46], which belongs to the Solute Carrier 22 (SLC22; organic cation/anion/zwitterion transporters) family and is expressed in the basolateral membrane of renal proximal tubule cells [47, 48]. OAT1 is one of major constituents of the renal organic anion transport pathway and plays a key role in the distribution and renal tubular secretion of a variety of endogenous and exogenous organic anions [48, 49]. In accordance with its crucial role in renal active secretion of many drugs, such as nonsteroidal anti-inflammatory drugs, antiviral drugs, antibiotics, diuretics, antineoplasmics, antiepileptics, OAT1-mediated DDIs have been observed clinically [50]. For example, when used with the known OAT1 inhibitor probenecid, the renal excretion of benzylpenicillin and ciprofloxacin was reduced [51].
Recent studies have demonstrated the inhibitory effect of individual compounds in TCM and how these can modulate the ability of OAT1 transporting its substrates [52–54]. Thus the possibility of OAT1-mediated TCM–drug interactions should be seriously considered when TCM are co-administrated with drugs that are transported by OAT1 [52, 55]. Each TCM usually consists of multiple compounds as a complex mixture and there are more than 600 TCMs that are currently used in clinical practice. Therefore, it is important to develop an effective and rapid screening approach for putative TCMs with OAT1-modulating activities. Computational modeling of drug–transporter interactions has showed great potential to identify DDIs mediated by various transporters, including OAT1 [56–58]. Since OAT1 is a transmembrane protein and there is no crystal structure available for structural details on ligand-protein interactions, we constructed a structural model of OAT1 for a molecular-docking study that was followed by the pharmacophore modeling technique [59] to predict whether TCMs reported in the Chinese Pharmacopoeia could interact with this renal drug uptake transporter. To our knowledge there have been no large-scale efforts to evaluate potential TCM interactions with OAT1. The goal of our current study was to use previously published OAT1 inhibitors, to build and test a pharmacophore model for OAT1 inhibition, which could be useful to facilitate identification of TCM-OAT1 interactions that could then be tested experimentally. Results from this preliminary study provide valuable information to understand and estimate OAT1-mediated TCM–drug interactions, which may possess important clinical relevance.
4.2.2 Construction of TCM Compound Database
Information about clinically relevant TCMs was collected from the Chinese Pharmaceutical Encyclopedia (2010 edition) [60]. Literature mining and database searching were then performed to identify small molecule compounds that have been described in these TCMs by the keyword-based approach. This process resulted in chemical structures of 10,914 compounds from 611 TCMs after removing inorganic compounds and metals. Two-dimensional (2D) and three-dimensional (3D) structural configurations of these compounds were generated using the Molecular Operating Environment (MOE) (Chemical Computing Group, Montreal, Canada) and saved in a database format for further computational evaluations. The TCM compound database was updated to remove counter-ions and corresponding protonation stage of the compounds was decided under pH value of 7.4. The 3D structure of each compound was constructed using the MMFF94x force field using MOE. Then the low energy conformers for each compound were generated using the conformation import methodology implemented in MOE.
4.2.3 OAT1 Inhibitor Pharmacophore Development
More than 50 compounds were collected from the UCSF-FDA TransPortal website [61] (http://bts.ucsf.edu/fdatransportal/) as known inhibitors of human OAT1 and used for pharmacophore generation. These compounds have been experimentally validated to inhibit para-aminohippuric acid (PAH) uptake via OAT1. Since the goal of this study was to construct a representative inhibitor pharmacophore model and apply it to identify OAT1 inhibitors from TCMs that may induce herb-drug interactions clinically, we considered potent inhibitors with IC50 less than 50 μM as OAT1 actives in this training set, while drugs with IC50 more than 50 μM were included as OAT1 inactives to derive unfavorable features for inhibition. This process led to 28 compounds (24 actives and 4 inactives) for inhibitor pharmacophore generation. To explore the conformational space of these OAT1 inhibitors, a fragment-based high-throughput conformer generation approach accessible as the “conformation import” method in MOE was used to generate conformation clusters for each molecule for further calculations.
Pharmacophores are the key features that are important for biological activity and their geometric arrangement in space. The initial OAT1 inhibitor pharmacophore model was developed using only five molecules from 24 inhibitors with the lowest IC50 data, including mefenamic acid, diclofenac, indomethacin, ketoprofen, and naproxen. The pharmacophore elucidation protocol in MOE was applied in this step. Atoms in the compounds were annotated according to the Unified Scheme of MOE pharmacophore searching application. The remaining less-active OAT1 inhibitors and inactives were then mapped to this model and their chemical and spatial features were incorporated into the model.
4.2.4 External Test Set Evaluation
Following the development of the OAT1 inhibitor pharmacophore model, we performed model validation by utilizing it as query for in silico screening of drugs in the NIH clinical collection (NCC) and NCC2 (Evotec, USA) whose OAT1 inhibitory activities have been determined experimentally [56]. The test set included 10 OAT1 actives and 8 OAT1 inactives. The predictive capability of our pharmacophore model was evaluated by identification of molecules with confirmed inhibitory activities from the NCC and NCC2 databases.
4.2.5 Database Searching
After model validation, the pharmacophore model was then used to search the TCM compound database. Hits exhibiting a good fit to the generated pharmacophore model were retrieved from our TCM compound database for further evaluation. The root mean square deviation between centroids of pharmacophore features and annotated points in the compounds were calculated to evaluate the quality of the pharmacophore mapping. PubMed (http://ncbi.nlm.nih.gov/pubmed/) was searched for the likely connection between OAT1 and these computationally identified TCM compounds or their original TCMs. Jarvis-Patrick clustering of TCM compounds mapped to OAT1 inhibitor pharmacophore was performed in MOE through 2D MACCS structural keys (MDL Ltd, CA, USA) and resulted in partitioning into 32 categories by (sub)structure similarity.
4.2.6 Results: OAT1 Inhibitor Pharmacophore
Twenty-four known OAT1 inhibitors were used to develop the OAT1 inhibitor pharmacophore model and the inhibitors had three common features: a negative ionizable center (F1), a hydrophobe (F2), and the third feature (F3) that can be an aromatic center or a hydrophobic centroid, along with six excluded volumes (Figure 4.1). This pharmacophore model successfully mapped 19 inhibitors among the 24 highly active OAT1 inhibitors (IC50 < 50 μM); however, it failed to identify two thiazide diuretic drugs (chlorothiazide and trichlormethiazide). This is apparently due to the absence of a negative anionic center in these compounds, which is the essential feature in our model. Recent results have showed that chlorothiazide may work as substrates of OAT1 but it is not clear whether this drug binds to the same site as PAH does [62]. Future work is planned to use these drugs to elucidate the important pharmacophoric features for OAT1 substrates and comparison of the substrate and inhibitor pharmacophores would highlight their chemical similarities/dissimilarities. The carboxyl group was present for the other three drugs missed by the model (adipate, α-ketoglutarate, and glutarate). However they all have small size in terms of volume, suggesting that this may be an important factor for the current OAT1 inhibitor pharmacophore.

Figure 4.1 An OAT1 inhibitor pharmacophore model that consists of a negative ionizable feature (F1, red), one hydrophobe (F2, yellow), and a third feature that can be an aromatic center or a hydrophobic centroid (F3, yellow). In addition, six excluded volumes shown as gray spheres were present in this model. A potent OAT1 inhibitor, bumetanide (IC50 = 6 μM), has been displayed with the model and the atoms are colored by atom type (carbon, gray; nitrogen, blue; oxygen, red; phosphorus, yellow).
4.2.7 Results: OAT1 Inhibitor Pharmacophore Evaluation
Duan et al. [56] identified 10 highly potent OAT1 inhibitors from clinical drug libraries and those drugs established the test set to evaluate our OAT1 inhibitor pharmacophore model (Table 4.1). Mefenamic acid and Ketoprofen were the only two common drugs in the training set and test set and these were excluded from analysis. Pharmacophore comparison on the remaining eight active molecules in the test set showed that seven were mapped to the current OAT1 inhibitor pharmacophore. The only drug missed was nitazoxanide, which does not possess a negatively charged moiety that appears essential. Furthermore, none of inactives (eight drugs) matched with the current OAT1 inhibitor model. These results attest to the specificity of the model. The same group that published inhibitory activities of the test set drugs also generated a non-discriminative inhibitor pharmacophore for OAT1 and OAT3, which partially overlapped with our model on one negative ionizable feature and an aromatic center. However, the inter-feature distances were varied between two models and our model included an additional feature that could be aromatic or hydrophobic. These two common features therefore appear most important for inhibitors interacting with OAT1. The distance between these two features was 4.1 Å for our model versus 5.7 Å in previous model and this distance variation may relate to the selection of different OAT1 substrates during the experiments, as a subtle difference in local receptor conformations can be induced in the same binding site by various substrates. Inhibitors were selected in this study when PAH was used as substrate for OAT1, while Duan et al. used 6-carboxyfluorescein to determine inhibitory activities of clinical drugs. We speculated that these two sets of inhibitors may interact with an overlapping binding site region of OAT1 based on shared common features.
Table 4.1 Representative molecules used for OAT1 inhibitor pharmacophore model generation and validation
| Molecules | Detailsa | References | |
| Training set | Mefenamic acid | Non-steroidal anti-inflammatory drug (IC50 = 1 μM) | [80] |
| Novobiocin | An aminocoumarin antibiotic (IC50 = 38 μM) | [81] | |
| Ketoprofen | A non-steroidal anti-inflammatory drug (IC50 = 4 μM) | [80] | |
| Fluvastatin | A hypolipidemic drug to hypercholesterolemia and to prevent cardiovascular disease (IC50 = 26 μM) | [82] | |
| Probenecid | A uricosuric drug to treat gout and hyperuricemia (IC50 = 6.5 μM) | [83] | |
| Test set | Meclofenamic acid | A non-steroidal anti-inflammatory drug | [56] |
| Ketorolac | A non-steroidal anti-inflammatory drug | ||
| Telmisartan | An angiotensin II receptor antagonist | ||
| Oxaprozin | A non-steroidal anti-inflammatory drug | ||
| Amlexanox | An anti-inflammatory and anti-allergic immunomodulator |
a Activities were reported against OAT1.
4.2.8 Results: TCM Compound Database Searching Using OAT1 Inhibitor Pharmacophore
Using our OAT1 inhibitor pharmacophore model as a query, we searched through the TCM compound database and mapped 144 compounds from over 10,000 compounds. The hit rate is around 1.3% and it showed that our model is rather selective against the large database. Since the same compounds may exist in different TCMs, there were 101 unique TCM compounds after removing the duplicates, which originate from 85 kinds of TCM (about 13% of all TCMs). For example, Rhein, the active compound in Rheum sp., is found in five other TCMs from our database. The majority of 85 TCMs only had one compound mapped to the model (Figure 4.2) and their potential to be involved in TCM–drug interactions probably would be insignificant considering the relative low quantity of a single compound in one TCM. About 11.5% (11/87) of TCMs mapped by our method included more than two compounds that were predicted as OAT1 inhibitors and detailed investigations would be valuable for their role in TCM–drug interactions (listed in Figure 4.2). A literature survey of these 11 TCMs showed that Radix Scutellariae, with three OAT1 inhibitors predicted by the model, inhibited the uptake of PAH through OAT1 about 51% by a single TCM preparation [63]. Among 11 selected TCMs, Aristolochia debilis is known for its nephrotoxicity that is related to uptake of compounds from this TCM by OATs [64] and three TCM compounds (aristolochic acids) in this herb were predicted to show strong interactions with OAT1, which is consistent with previous experimental data [65].
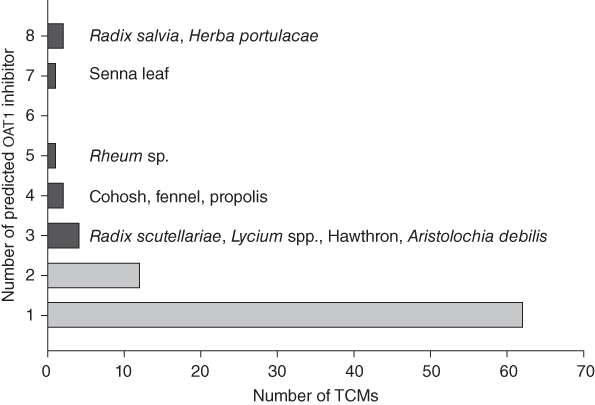
Figure 4.2 The distribution of predicted TCM compounds with OAT1 inhibitory activity in medicinal TCMs. The black bars represent TCMs with three or more compounds mapped to the pharmacophore model and the names of these TCMs are listed in the figure.
A further PubMed search was performed for several predicted TCM compounds to confirm their direct inhibitory activities against OAT1. In agreement with results from pharmacophore searching, components of Radix Salvia, salvianolic acids, lithospermic acid, and rosmarinic acid, have all been demonstrated to competitively inhibit OAT1 experimentally [66]. Rhein, found in two of the selected TCMs (Rheum sp. and sennae leaf), is the major constituent of these two TCMs and displayed a potent nanomolar activity for OAT1 in vitro and a calculated DDI index of 5.0 [54], suggesting high probability of occurrence of herb-drug interactions when taken with drugs that are transported by OAT1. In this study, several cinnamic acids, such as ferulic acid and sinapinic acid, were predicted to inhibit OAT1 activities by the pharmacophore model, which was also consistent with published results [52]. Table 4.2 summarizes the information for these TCM compounds and Figure 4.3a-h shows how these compounds matched to the common features of OAT1 inhibitor pharmacophore. Isoferulic acid, a structural analog of ferulic acid, was also selected by our pharmacophore and we expected this compound would inhibit OAT1 because of great structural similarity to ferulic acid (Table 4.3). These cinnamic acids exist in five selected TCMs; however, their involvement in drug interactions needs more experimental investigation.
Table 4.2 Example TCM compounds with experimental information about interactions with OAT1.
| TCM compound | Chemical structure | IC50 (μM) | DDI indexa | References |
| Rhein |

|
0.077 | 5.0 | [54] |
| Aristolochic acid I |
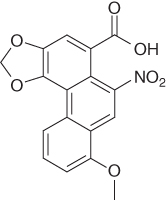
|
0.84b | 0.19 | [84] |
| Salvianolic acid A |

|
5.6b | c | [52] |
| Lithospermic acid |

|
20.8b | 0.01 | [52] |
| Rosmarinic acid |

|
0.32b | 1.6 | [52] |
| Sinapinic acid |

|
11.02 | c | [53] |
| Ferulic acid |

|
9.01 | 0.03 | [53] |
a DDI index were calculated using previously reported data.
b Ki values were reported in the corresponding references.
c Calculation was not done owing to lack of information.

Figure 4.3 TCM compounds mapping to the OAT1 inhibitor pharmacophore. The pharmacophore consists of a negative ionizable feature (red) and two hydrophobic features (yellow). For clarity, the excluded volumes are not shown here. (a) rhein; (b) aristolochic acid I; (c) salvianolic acid A; (d) lithospermic acid; (e) rosmarinic acid; (f) ferulic acid; (g) sinapinic acid; (h) and isoferulic acid.
Table 4.3 Structurally similar TCM compounds without experimental validation
| TCM compound | Chemical structure | Validated compound | Tsa |
| Isoferulic acid |

|
Ferulic acid | 1.0 |
| Cimicifugic acid |

|
Lithospermic acid | 0.77 |
| Lonchocarpric acid |

|
Lithospermic acid | 0.59 |
a Tanimoto score (Ts) using 2D MACCS fingerprint.
4.2.9 Discussion
TCM has continuously displayed beneficial therapeutic effects against various diseases in the clinic, including cardiovascular diseases, infectious diseases, and cancers [67]. As a form of complementary/alternative medicine, it is accepted in Western countries, such as the United States, and a recent survey [36] suggested that 20% of the population reported the use of herbal medicines that make up 80% of TCMs. However, compared to its broad clinical applications, pharmacological studies on TCM are limited, particularly on their potential toxicological or adverse effects. For example, there are extremely limited studies on TCM–drug interactions when used concomitantly. Some clinical evidence is available to support the existence of TCM–drug interactions [68, 69]. Most of the attention has been focused on how TCMs disrupt activities of drug-metabolizing enzymes such as cytochrome P450 [70]. Despite the important role of transporters in drug absorption and disposition which has been well recognized, information about TCM-transporter interactions is sparse and as most of the studies have concentrated on P-glycoprotein [71]. Few studies have been conducted to assess the ability of TCMs as renal transporter substrates or inhibitors, which clearly is a possible mechanism for TCM–drug interactions. Therefore, it is critical to characterize the level of TCM-renal transporter associations in order to prevent unexpected drug toxicity when TCM and drugs are administrated concomitantly.
For synthetic drugs, experimental screening of their capabilities to modulate OAT1 activity has been performed to prevent potential DDIs [56]. However, this approach is probably not feasible to screen TCM compounds owing to the enormous number of compounds to be tested. Prioritization for experimental testing would be required to speed up the process and gain more valuable information on TCM-transporter interactions. Computational modeling in combination with in vitro experiments has been used to enrich our knowledge about substrates and inhibitors of renal transporters [72, 73], which also would provide useful information about TCM-renal transporter interactions. We present such an approach aimed at the identification of OAT1 inhibitors from TCM by building an OAT1 inhibitor pharmacophore model using previously published in vitro data on inhibitors of this transporter. To measure model accuracy, additional literature information was used to form a validation set with experimental observations that could be compared with predicted results. A further literature search confirmed an important relationship between compounds in medicinal TCMs and OAT1 suggested by the current computational model, which may possess significant clinical implications by impacting pharmacokinetics or pharmacodynamics of drugs co-administrated with TCMs.
In vitro experiments have shown that the same drug displayed altered levels of inhibition when different substrates of one transporter were present, suggesting that inhibitors may interact with the transporter through diverse patterns [61]. This observation makes it difficult to generate one comprehensive pharmacophore hypothesis to map all known inhibitors. Here we are interested in potent OAT1 inhibitors from TCMs, which then may show clinically relevant interactions with other drugs; thus a highly specific pharmacophore model would be preferred. To establish a single OAT1 inhibitor pharmacophore model, it is important to select inhibitors whose OAT1 activities are evaluated by inhibiting uptake of the same substrate, therefore focusing on explaining a unique OAT1 inhibitory mechanism. In this study, the OAT1 substrate is PAH, which is commonly used to measure renal plasma flow due to its primary secretion by renal tubules. Although the PAH-dependent OAT1 inhibitor pharmacophore model only accounts for one specific inhibition mechanism, it would represent a large portion of OAT1 inhibitors and display high specificity to the PAH binding site of OAT1.
Using 24 highly active inhibitors that disrupt the ability of OAT1 to transport PAH, a three-feature pharmacophore model was constructed to represent important structural elements of OAT1 inhibitors. We found that the negative ionizable feature was important for potent OAT1 inhibitors. However, several drugs without this feature, such as thiazide diuretics, also strongly blocked transport of the OAT1 substrate, suggesting they may interact with the transporter through a different inhibitory mode and a separate pharmacophore will be needed to identify OAT1 inhibitors functioning similarly to those drugs. Furthermore, comparison with another published OAT1 inhibitor pharmacophore [56] revealed two overlapping features (one negative center and an aromatic center) that possibly are required for inhibitor binding. In our model, one additional feature (F3) suggested a minimal size threshold for OAT1 inhibitors by an approximate 8 Å separation between F1 and F3. During the model evaluation step, the current model successfully mapped seven out of eight OAT1 active drugs from an external test set and showed great discrimination between actives and inactives, indicating that the model can identify potent inhibitors that are not in the training set. OAT1 substrate pharmacophores with common features have been constructed to identify metabolites transported by OAT1 [57, 74]. Comparison with these models again highlighted the importance of the anionic center, which appears in both inhibitor and substrate models. In addition to pharmacophore modeling, quantitative structure-activity relationships have been developed for a series of antiviral compounds and mouse OAT1 inhibitors showed higher polar surface areas relative to those for mouse OAT3 and OAT6 [75]. This information is also valuable for computational screening for OAT1 inhibitors.
There have been no previous database screening efforts with TCM compounds against OAT1 on a large scale. The current effort therefore presents a first attempt to perform this task using our OAT1 inhibitor pharmacophore and a very small percentage of TCM compounds; 144 TCM compounds matching the key features of pharmacophore model were identified by our model as potential OAT1 inhibitors and retrieved from a large database with over 10,000 compounds. This high selectivity may partially relate to the PAH-specific OAT1 inhibitor pharmacophore we constructed. Several TCM compounds with reported OAT1 inhibitory activities were also revealed by our method (shown in Table 4.2), such as Rhein with nanomolar potency. The OAT1 activities remain to be experimentally validated for a large portion of TCM compounds identified in this work. Chemical similarity analysis indicated that 144 TCM compounds belonged to 32 distinct structural clusters and more than half of the predicted compounds (68 compounds) can be grouped into two clusters (data not shown). All TCM compounds with previously known OAT1 activities (shown in Table 4.2) were found in these two clusters except Rhein, which was located in a separate cluster. These results showed predicted TCM compounds in these two clusters shared good structural similarities and they very likely modulate OAT1 in a similar manner as those confirmed TCM compounds.
On the basis of the number of predicted OAT1 inhibitors in each TCM, 11 TCMs were selected and investigated as potential perpetrators for OAT1-mediated TCM–drug interactions. Additional literature search revealed experimental evidence to support the associations with OAT1 for 5 out of these 11 TCMs (Salvia miltiorrhizae, A. debilis, Scutellaria baicalensis, senna leaf, and Rheum sp.). The current pharmacophore model also mapped hydroxycinnamic acid, a class of aromatic acid or phenylpropanoids with a C6−C3 skeleton, such as ferulic acid and sinapinic acid, to common features of potent OAT1 inhibitors. These organic acids are found in five unconfirmed TCMs (cohosh, hawthorn, fennel, propolis, and Herba portulacae). Table 4.3 lists the Tanimoto scores of three TCM compounds from these unconfirmed TCMs with validated compounds, which suggests that these unconfirmed compounds share good structural similarity with the validated compounds. It is possible they also would inhibit OAT1 activity. Since the quantitative content of these acids in TCM are not available, experiments are warranted to prove the participation of interactions with OAT1 for these TCMs. Notably, all these five TCMs can be used as dietary material, which suggests that they could be consumed in large quantities by people. For example, black cohosh is a popular dietary supplement among women for management of menopausal symptoms. The extract of this plant has been shown to moderately inhibit the transport of estrone-3-sulfate uptake by another organic anion transporter, OATP-B [76]. Clearly, according to the previous report and our prediction, it is worth exploring the potential OAT1 inhibitory activity of black cohosh. Lycium spp., a well-known culinary fruit, which is another TCM selected by our method and estimated to be involved in OAT1-mediated TCM–drug interactions in this study. Although there is no information about OAT1 activity of Lycium spp., a clinical-evidence-based evaluation indicated that this fruit can induce interactions with warfarin [77, 78]. More experimental testing is required to demonstrate the interactions between Lycium spp. and OAT1 and the possibility of disrupting OAT1 substrate drug uptake by this TCM.
Renal active secretion of drugs would involve both transporter-mediated basolateral uptake and apical efflux. In this study, we focused on the potential influence of TCM on the OAT1-mediated basolateral uptake, which is a portion of the complex dynamics of drug movement across the renal membrane. To gain a more complete understanding of the potential of TCM–drug interactions, it is important to characterize the contribution from other renal transporters, including another important uptake transporter OAT3 and the apical efflux transporter (multidrug resistance protein 4) [79]. More studies on interactions between TCM and these transporters are currently ongoing to clarify the mechanism of transporter-mediated TCM–drug interactions in kidney.
4.3 Conclusion
In conclusion, we developed an OAT1 inhibitor pharmacophore to estimate inhibitory activity of TCMs on this important renal transporter, which might potentially induce OAT1-related TCM–drug interactions and lead to unexpected renal accumulation of concomitantly used OAT1 substrate drugs. Eleven TCMs were predicted to inhibit OAT-mediated substrate uptake and five of these were subsequently validated by reported experimental data. The computational pharmacophore approach used in the study could be useful in evaluating modulation of other drug transporters by TCMs. The results of the study should provide helpful information related to drug interactions in TCM safety research. Furthermore, our results can be used as future references for general public and clinical practitioners about the potential risk of combinational usage of TCM and drugs, as well as helping to make related regulatory policies.
Acknowledgment
The authors acknowledge the funding on this work from National Youth Top-notch Talent Support Program and the National Natural Science Foundation of China (No. 81173465).
References
- 1 Wojcikowski, K., Johnson, D.W., and Gobe, G. (2004) Medicinal herbal extracts – renal friend or foe? Part one: the toxicities of medicinal herbs. Nephrology, 9, 313–318.
- 2 Sato, A., Toyoshima, M., Kondo, A. et al. (1997) Pneumonitis induced by the herbal medicine Sho-saiko-to in Japan. Nihon Kyobu Shikkan Gakkai Zasshi, 35, 391–395.
- 3 Guo, Y.J., Wang, D.W., Meng, L., and Wang, Y.Q. (2015) Analysis of anaphylactic shock caused by 17 types of traditional Chinese medicine injections used to treat cardiovascular and cerebrovascular diseases. BioMed. Res. Int., 2015, 420607.
- 4 Cardenas, A., Restrepo, J.C., Sierra, F., and Correa, G. (2006) Acute hepatitis due to shen-min: a herbal product derived from Polygonum multiflorum. J. Clin. Gastroenterol., 40, 629–632.
- 5 Mazzanti, G., Battinelli, L., Daniele, C. et al. (2004) New case of acute hepatitis following the consumption of Shou Wu Pian, a Chinese herbal product derived from Polygonum multiflorum. Ann. Intern. Med., 140, W30.
- 6 Park, G.J., Mann, S.P., and Ngu, M.C. (2001) Acute hepatitis induced by Shou-Wu-Pian, a herbal product derived from Polygonum multiflorum. J. Gastroenterol. Hepatol., 16, 115–117.
- 7 Committee of Chinese Pharmcopeia (2015) Chinese Pharmacopeia, Chinese Medical & Science Press, Beijing.
- 8 Ekins, S. (2007) Computational Toxicology: Risk Assessment for Pharmaceutical and Environmental Chemicals, Wiley-Interscience, United States.
- 9 Devarbhavi, H. (2012) An update on drug-induced liver injury. J. Clin. Exp. Hepatol., 2, 247–259.
- 10 Regev, A. (2014) Drug-induced liver injury and drug development: industry perspective. Semin. Liver Dis., 34, 227–239.
- 11 Huang, S.H., Tung, C.W., Fulop, F., and Li, J.H. (2015) Developing a QSAR model for hepatotoxicity screening of the active compounds in traditional Chinese medicines. Food Chem. Toxicol., 78, 71–77.
- 12 Ye, L.W., Zhu, Y., Jin, R. et al. (2014) Predicting hepatotoxicity of compounds from traditional Chinese medicines using tree models. Chin. Pharm. J., 49 (18), 1583–1588.
- 13 Liu, H., Li, T., Chen, L. et al. (2016) To set up a logistic regression prediction model for hepatotoxicity of Chinese herbal medicines based on traditional Chinese medicine theory. Evid. Based Complement. Alternat. Med.: eCAM., 2016, 7273940.
- 14 Wang XZ, Zhu, YL, Jin, RM, LIU, JG, et al.. Predicting nephrotoxicity of drugs using mathematical models. Chin. J. New Drugs 2014;23(13), 1565–1568.
- 15 Zhang, J.F., Jiang, L.D., and Zhang, Y.L. (2015) Application of support vector machine approach in studying nephron toxicity of Chinese medicinal materialsZhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi = . China J. Chinese Mater. Med., 40, 1134–1138.
- 16 Lei, L., Wang, X., Zhang, L. et al. (2015) QSAR study on rat cardiotoxicity of chemical component of Chinese herbs. World Sci. Technol./Modern. Trad. Chinese Med. Mater. Med., 17, 5.
- 17 Zhang, J.F., Jiang, L.D., and Zhang, Y.L. (2014) Application of support vector machine in screening neurotoxic compounds from traditional Chinese medicineZhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi =. China J. Chinese Mater. Med., 39, 3330–3334.
- 18 Ai, N., Fan, X., and Ekins, S. (2015) In silico methods for predicting drug–drug interactions with cytochrome P-450s, transporters and beyond. Adv. Drug Deliv. Rev., 86, 46–60.
- 19 Shao, X., Ai, N., Xu, D., and Fan, X. (2016) Exploring the interaction between Salvia miltiorrhiza and human serum albumin: Insights from herb-drug interaction reports, computational analysis and experimental studies. Spectrochim. Acta A, Mol. Biomol. Spectrosc., 161, 1–7.
- 20 Su, Z., Zhang, B., Zhu, W., and Du, Z. (2012) In silico and in vivo evaluation of flavonoid extracts on CYP2D6-mediated herb–drug interaction. J. Mol. Model., 18, 4657–4663.
- 21 Hu, T., Zhou, X., Wang, L. et al. (2015) Effects of tanshinones from Salvia miltiorrhiza on CYP2C19 activity in human liver microsomes: enzyme kinetic and molecular docking studies. Chem. Biol. Interact., 230, 1–8.
- 22 Cui, Z., Kang, H., Tang, K. et al. (2015) Screening ingredients from herbs against Pregnane X receptor in the study of inductive herb–drug interactions: combining pharmacophore and docking-based rank aggregation. BioMed. Res. Int., 2015, 657159.
- 23 Xu, M., Dong, P., Tian, X. et al. (2016) Drug interaction study of natural steroids from herbs specifically toward human UDP-glucuronosyltransferase (UGT) 1A4 and their quantitative structure activity relationship (QSAR) analysis for prediction. Pharmacol. Res., 110, 139–150.
- 24 Cao, D., Liu, S., Fan, L., and Liang, Y. (2014) QSAR analysis of the effects of OATP1B1 transporter by structurally diverse natural products using a particle swarm optimization-combined multiple linear regression approach. Chemometr. Intell. Lab. Syst., 130, 7.
- 25 Abdel-Rahman, S.M., Marcucci, K., Boge, T. et al. (1999) Potent inhibition of cytochrome P-450 2D6-mediated dextromethorphan O-demethylation by terbinafine. Drug Metab. Dispos., 27, 770–775.
- 26 Fisel, P., Renner, O., Nies, A.T. et al. (2014) Solute carrier transporter and drug-related nephrotoxicity: the impact of proximal tubule cell models for preclinical research. Expert Opin. Drug Metab. Toxicol., 10, 395–408.
- 27 Maeda, K. and Sugiyama, Y. (2013) Transporter biology in drug approval: Regulatory aspects. Mol. Aspects Med., 34, 711–718.
- 28 Noguchi, K., Katayama, K., and Sugimoto, Y. (2014) Human ABC transporter ABCG2/BCRP expression in chemoresistance: basic and clinical perspectives for molecular cancer therapeutics. Pharmacogenomics Pers. Med., 7, 53–64.
- 29 Causevic-Ramosevac, A. and Semiz, S. (2013) Drug interactions with statins. Acta Pharm., 63, 277–293.
- 30 Ieiri, I. (2012) Functional significance of genetic polymorphisms in P-glycoprotein (MDR1, ABCB1) and breast cancer resistance protein (BCRP, ABCG2). Drug Metab. Pharmacokinet., 27, 85–105.
- 31 Koepsell, H. (2013) The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Aspects Med., 34, 413–435.
- 32 Wang, L. and Sweet, D.H. (2013) Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J., 15, 53–69.
- 33 Qi, F.H., Wang, Z.X., Cai, P.P. et al. (2013) Traditional Chinese medicine and related active compounds: a review of their role on hepatitis B virus infection. Drug Discov. Therap., 7, 212–224.
- 34 Guo, M., Liu, Y., Gao, Z.Y., and Shi, D.Z. (2014) Chinese herbal medicine on dyslipidemia: progress and perspective. Evid. Based Complementary Altern. Med.: eCAM, 2014, 163036.
- 35 Tzeng, T.F., Liou, S.S., and Liu, I.M. (2013) The selected traditional Chinese medicinal formulas for treating diabetic nephropathy: perspective of modern science. J. Tradit. Complement. Med., 3, 152–158.
- 36 Bent, S. (2008) Herbal medicine in the United States: review of efficacy, safety, and regulation: grand rounds at University of California, San Francisco Medical Center. J. Gen. Intern. Med., 23, 854–859.
- 37 Interactions with herbal products: what do we know? Drug Therap. Bull. 2014;52:18–21.
- 38 Ulbricht, C., Chao, W., Costa, D. et al. (2008) Clinical evidence of herb–drug interactions: a systematic review by the natural standard research collaboration. Curr. Drug Metab., 9, 1063–1120.
- 39 Marchetti, S., Mazzanti, R., Beijnen, J.H., and Schellens, J.H. (2007) Concise review: clinical relevance of drug drug and herb drug interactions mediated by the ABC transporter ABCB1 (MDR1, P-glycoprotein). Oncologist, 12, 927–941.
- 40 Hu, Z., Yang, X., Ho, P.C. et al. (2005) Herb–drug interactions: a literature review. Drugs, 65, 1239–1282.
- 41 Li, X.P., Zhang, C.L., Gao, P. et al. (2013) Effects of andrographolide on the pharmacokinetics of aminophylline and doxofylline in rats. Drug Res., 63, 258–262.
- 42 Xue, X.P., Qin, X.L., Xu, C. et al. (2013) Effect of Wuzhi tablet (Schisandra sphenanthera extract) on the pharmacokinetics of cyclosporin A in rats. Phytother. Res., 27, 1255–1259.
- 43 Chan, E., Tan, M., Xin, J. et al. (2010) Interactions between traditional Chinese medicines and Western therapeutics. Curr. Opin. Drug Discov. Dev., 13, 50–65.
- 44 Lepist, E.I. and Ray, A.S. (2012) Renal drug–drug interactions: what we have learned and where we are going. Expert Opin. Drug Metab. Toxicol., 8, 433–448.
- 45 Wolff, N.A., Werner, A., Burkhardt, S., and Burckhardt, G. (1997) Expression cloning and characterization of a renal organic anion transporter from winter flounder. FEBS Lett., 417, 287–291.
- 46 Sekine, T., Watanabe, N., Hosoyamada, M. et al. (1997) Expression cloning and characterization of a novel multispecific organic anion transporter. J. Biol. Chem., 272, 18526–18529.
- 47 Sweet, D.H., Wolff, N.A., and Pritchard, J.B. (1997) Expression cloning and characterization of ROAT1. The basolateral organic anion transporter in rat kidney. J. Biol. Chem., 272, 30088–30095.
- 48 Miyazaki, H., Sekine, T., and Endou, H. (2004) The multispecific organic anion transporter family: properties and pharmacological significance. Trends Pharmacol. Sci., 25, 654–662.
- 49 Rizwan, A.N. and Burckhardt, G. (2007) Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm. Res., 24, 450–470.
- 50 Uwai, Y., Taniguchi, R., Motohashi, H. et al. (2004) Methotrexate–loxoprofen interaction: involvement of human organic anion transporters hOAT1 and hOAT3. Drug Metab. Pharmacokinet., 19, 369–374.
- 51 Jaehde, U., Sorgel, F., Reiter, A. et al. (1995) Effect of probenecid on the distribution and elimination of ciprofloxacin in humans. Clin. Pharmacol. Therap., 58, 532–541.
- 52 Wang, L. and Sweet, D.H. (2013) Interaction of natural dietary and herbal anionic compounds and flavonoids with human organic anion transporters 1 (SLC22A6), 3 (SLC22A8), and 4 (SLC22A11). Evid. Based Complement. Altern. Med.: eCAM, 2013, 612527.
- 53 Wang, L. and Sweet, D.H. (2012) Potential for food-drug interactions by dietary phenolic acids on human organic anion transporters 1 (SLC22A6), 3 (SLC22A8), and 4 (SLC22A11). Biochem. Pharmacol., 84, 1088–1095.
- 54 Wang, L., Pan, X., and Sweet, D.H. (2013) The anthraquinone drug rhein potently interferes with organic anion transporter-mediated renal elimination. Biochem. Pharmacol., 86, 991–996.
- 55 Uwai, Y., Ozeki, Y., Isaka, T. et al. (2011) Inhibitory effect of caffeic acid on human organic anion transporters hOAT1 and hOAT3: a novel candidate for food–drug interaction. Drug Metab. Pharmacokinet., 26, 486–493.
- 56 Duan, P., Li, S., Ai, N. et al. (2012) Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: discovery and molecular characterization. Mol. Pharm., 9, 3340–3346.
- 57 Kouznetsova, V.L., Tsigelny, I.F., Nagle, M.A., and Nigam, S.K. (2011) Elucidation of common pharmacophores from analysis of targeted metabolites transported by the multispecific drug transporter–Organic anion transporter1 (Oat1). Bioorg. Med. Chem., 19, 3320–3340.
- 58 Ahn, S.Y., Eraly, S.A., Tsigelny, I., and Nigam, S.K. (2009) Interaction of organic cations with organic anion transporters. J. Biol. Chem., 284, 31422–31430.
- 59 Guner, O.F. and Bowen, J.P. (2013) Pharmacophore modeling for ADME. Curr. Top. Med. Chem., 13, 1327–1342.
- 60 Committee, N.P. (2010) ChinesePharmacopoeia, Medicine Science and Technology Press of China, Beijing.
- 61 Morrissey, K.M., Wen, C.C., Johns, S.J. et al. (2012) The UCSF-FDA TransPortal: a public drug transporter database. Clin. Pharmacol. Therap., 92, 545–546.
- 62 Juhasz, V., Beery, E., Nagy, Z. et al. (2013) Chlorothiazide is a substrate for the human uptake transporters OAT1 and OAT3. J. Pharm. Sci., 102, 1683–1687.
- 63 Lin, C.C., Fan, H.Y., Kuo, C.W., and Pao, L.H. (2012) Evaluation of chinese-herbal-medicine-induced herb–drug interactions: focusing on organic anion transporter 1. Evid. Based Complementary Altern. Med.: eCAM, 2012, 967182.
- 64 Lebeau, C., Debelle, F.D., Arlt, V.M. et al. (2005) Early proximal tubule injury in experimental aristolochic acid nephropathy: functional and histological studies. Nephrol. Dial. Transplant., 20, 2321–2332.
- 65 Babu, E., Takeda, M., Nishida, R. et al. (2010) Interactions of human organic anion transporters with aristolochic acids. J. Pharm. Sci., 113, 192–196.
- 66 Zhang, J., Pan, X., Wang, C. et al. (2012) Pharmacophore modeling, 3D-QSAR studies, and in-silico ADME prediction of pyrrolidine derivatives as neuraminidase inhibitors. Chem. Biol. Drug Des., 79, 353–359.
- 67 Robinson, M.M. and Zhang, X. (2011) World Medicines Situation 2011, Traditional Medicines: Global Situation, Issues and Challenges, WHO.
- 68 Shi, S. and Klotz, U. (2012) Drug interactions with herbal medicines. Clin. Pharmacokinet., 51, 77–104.
- 69 Coxeter, P.D., McLachlan, A.J., Duke, C.C., and Roufogalis, B.D. (2004) Herb–drug interactions: an evidence based approach. Curr. Med. Chem., 11, 1513–1525.
- 70 Wu, J.J., Ai, C.Z., Liu, Y. et al. (2012) Interactions between phytochemicals from traditional Chinese medicines and human cytochrome P450 enzymes. Curr. Drug Metab., 13, 599–614.
- 71 Eichhorn, T. and Efferth, T. (2012) P-glycoprotein and its inhibition in tumors by phytochemicals derived from Chinese herbs. J. Ethnopharmacol., 141, 557–570.
- 72 Kido, Y., Matsson, P., and Giacomini, K.M. (2011) Profiling of a prescription drug library for potential renal drug–drug interactions mediated by the organic cation transporter 2. J. Med. Chem., 54, 4548–4558.
- 73 Nigam, S.K., Bush, K.T., and Bhatnagar, V. (2007) Drug and toxicant handling by the OAT organic anion transporters in the kidney and other tissues. Nat. Clin. Pract. Nephrol., 3, 443–448.
- 74 Wikoff, W.R., Nagle, M.A., Kouznetsova, V.L. et al. (2011) Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J. Proteome Res., 10, 2842–2851.
- 75 Truong, D.M., Kaler, G., Khandelwal, A. et al. (2008) Multi-level analysis of organic anion transporters 1, 3, and 6 reveals major differences in structural determinants of antiviral discrimination. J. Biol. Chem., 283, 8654–8663.
- 76 Fuchikami, H., Satoh, H., Tsujimoto, M. et al. (2006) Effects of herbal extracts on the function of human organic anion-transporting polypeptide OATP-B. Drug Metab. Dispos., 34, 577–582.
- 77 Rivera, C.A., Ferro, C.L., Bursua, A.J., and Gerber, B.S. (2012) Probable interaction between Lycium barbarum (Goji) and Warfarin. Pharmacotherapy, 32 (3), e50–e53.
- 78 Leung, H., Hung, A., Hui, A.C., and Chan, T.Y. (2008) Warfarin overdose due to the possible effects of Lycium barbarum L. Food Chem. Toxicol., 46, 1860–1862.
- 79 Smeets, P.H., van Aubel, R.A., Wouterse, A.C. et al. (2004) Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J. Am. Soc. Nephrol., 15, 2828–2835.