
 CHAPTER 16
CHAPTER 16
Metaheuristics in Bioinformatics: DNA Sequencing and Reconstruction
C. COTTA, A. J. FERNÁNDEZ, J. E. GALLARDO, G. LUQUE, and E. ALBA
Universidad de Málaga, Spain
16.1 INTRODUCTION
In recent decades, advances in the fields of molecular biology and genomic technologies have led to a very important growth in the biological information generated by the scientific community. The needs of biologists to utilize, interpret, and analyze that large amount of data have increased the importance of bioinformatics [1]. This area is an interdisciplinary field involving biology, computer science, mathematics, and statistics for achieving faster and better methods in those tasks.
Most bioinformatic tasks are formulated as difficult combinatorial problems. Thus, in most cases it is not feasible to solve large instances using exact techniques such as branch and bound. As a consequence, the use of metaheuristics and other approximate techniques is mandatory. In short, a metaheuristic [2,3] can be defined as a top-level general strategy that guides other heuristics to search for good solutions. Up to now there has been no commonly accepted definition for the term metaheuristic. It is just in the last few years that some researchers in the field have proposed a definition. Some fundamental characteristics:
- The goal is efficient exploration of the search space to find (nearly) optimal solutions.
- Metaheuristic algorithms are usually nondeterministic.
- They may incorporate mechanisms to avoid getting trapped in confined areas of the search space.
- The basic concepts of metaheuristics permit an abstract-level description.
- Metaheuristics are not problem specific.
- Metaheuristics usually allow an easy parallel implementation.
- Metaheuristics must make use of domain-specific knowledge in the form of heuristics that are controlled by the upper-level strategy.
The main advantages of using metaheuristics to solve bioinformatics tasks are the following:
- Problems of bioinformatics seldom require us to find an optimal solution. In fact, they require robust, nearly optimal solutions in a short time.
- Data obtained from laboratories inherently involve errors. Due to their non-deterministic process, metaheuristics are more tolerant in these cases than deterministic processes.
- Several tasks in bioinformatics involve the optimization of different objectives, thereby making the application of (population-based) metaheuristics more natural and appropriate.
In this chapter we first present a brief survey of metaheuristic techniques and the principal bioinformatic tasks. Later we describe in more detail two important problems in the area of sequence analysis: the DNA fragment assembly and the shortest common supersequence problem. We use them to exemplify how metaheuristics can be used to solve difficult bioinformatic tasks.
16.2 METAHEURISTICS AND BIOINFORMATICS
In this section we present some background information about metaheuristics and problems of bioinformatics.
16.2.1 Metaheuristics
As we said before, a metaheuristic [2,3] can be defined as a top-level general strategy that guides other heuristics to search for good solutions. There are different ways to classify and describe metaheuristic algorithms. One of them classifies them depending on the number of solutions: population based (a set of solutions) and trajectory based (work with a single solution). The latter starts with a single initial solution. At each step of the search the current solution is replaced by another (often the best) solution found in its neighborhood. Very often, such a metaheuristic allows us to find a local optimal solution and so are called exploitation-oriented methods. On the other hand, the former make use of a population of solutions. The initial population is enhanced through a natural evolution process. At each generation of the process, the entire population or a part of the population is replaced by newly generated individuals (often, the best ones). Population-based methods are often called exploration-oriented methods. In the next paragraph we discuss the features of the most important metaheuristics.
Trajectory-Based Metaheuristics
1. Simulated annealing (SA) [4] is a stochastic search method in which at each step, the current solution is replaced by another one selected randomly from the neighborhood. SA uses a control parameter, temperature, to determine the probability of accepting nonimproving solutions. The objective is to escape from local optima and so to delay the convergence. The temperature is gradually decreased according to a cooling schedule such that few nonimproving solutions are accepted at the end of the search.
2. Tabu search (TS) [5] manages a memory of solutions or moves used recently, called the tabu list. When a local optimum is reached, the search carries on by selecting a candidate worse than the current solution. To avoid the selection of the previous solution again, and so to avoid cycles, TS discards the neighboring candidates that have been used previously.
3. The basic idea behind variable neighborhood search (VNS) [6] is to explore successively a set of predefined neighborhoods to provide a better solution. It uses the descent method to get the local minimum. Then it explores either at random or systematically the set of neighborhoods. At each step, an initial solution is shaken from the current neighborhood. The current solution is replaced by a new one if and only if a better solution has been found. The exploration is thus restarted from that solution in the first neighborhood. If no better solution is found, the algorithm moves to the next neighborhood, generates a new solution randomly, and attempts to improve it.
Population-Based Metaheuristics
1. Evolutionary algorithms (broadly called EAs) [7] are stochastic search techniques that have been applied successfully in many real and complex applications (epistatic, multimodal, multiobjective, and highly constrained problems). Their success in solving difficult optimization tasks has promoted research in the field known as evolutionary computing (EC) [3]. An EA is an iterative technique that applies stochastic operators to a pool of individuals (the population). Every individual in the population is the encoded version of a tentative solution. Initially, this population is generated randomly. An evaluation function associates a fitness value to every individual, indicating its suitability to the problem. There exist several well-accepted subclasses of EAs depending on representation of the individuals or how each step of the algorithm is performed. The main subclasses of EAs are the genetic algorithm (GA), evolutionary programming (EP), the evolution strategy (ES), and some others not shown here.
2. Ant colony optimization (ACO) [8] have been inspired by colonies of real ants, which deposit a chemical substance (called a pheromone) on the ground. This substance influences the choices they make: The larger the amount of pheromone on a particular path; the larger the probability that an ant selects the path. In this type of algorithm, artificial ants are stochastic construction procedures that probabilistically build a solution by iteratively adding problem components to partial solutions by taking into account (1) heuristic information from the problem instances being solved, if available, and (2) pheromone trails that change dynamically at runtime to reflect the search experience acquired.
3. Scatter search (SS) [9] is a population-based metaheuristic that combines solutions selected from a reference set to build others. The method starts by generating an initial population of disperse and good solutions. The reference set is then constructed by selecting good representative solutions from the population. The solutions selected are combined to provide starting solutions to an improvement procedure. According to the result of such a procedure, the reference set and even the population of solutions can be updated. The process is iterated until a stopping criterion is satisfied. The SS approach involves different procedures, allowing us to generate the initial population, to build and update the reference set, to combine the solutions of such a set, to improve the solutions constructed, and so on.
Both approaches—trajectory-based and population-based—can also be combined to yield more powerful optimization techniques. This is particularly the case for memetic algorithms (MAs) [10], which blend ideas of different metaheuristics within the framework of population-based techniques. This can be done in a variety of ways, but most common approaches rely on the embedding of a trajectory-based technique within an EA-like algorithm (see refs. 11 and 12). It is also worth mentioning those metaheuristics included in the swarm intelligence paradigm, such as particle swarm optimization (PSO). These techniques regard optimization as an emergent phenomenon from the interaction of simple search agents.
16.2.2 Bioinformatic Tasks
In this subsection we describe the main bioinformatic tasks, giving in Table 16.1 the metaheuristic approaches applied to solve them. Based on the availability of the date and goals, we can classify the problems of bioinformatics as follows:
Alignment and Comparison of Genome and Proteome Sequences
From the biological point of view, sequence comparison is motivated by the fact that all living organism are related by evolution. This implies that the genes of species that are closer to each other should exhibit similarities at the DNA level. In biology, the sequences to be compared are either nucleotides (DNA, RNA) or amino acids (proteins). In the case of nucleotides, one usually aligns identical nucleotide symbols. When dealing with amino acids, the alignment of two amino acids occurs if they are identical or if one can be derived from the other by substitutions that are likely to occur in nature. A comparison of sequences comprises pairwise and simultaneous multiple sequence comparisons (and alignments). Therefore, algorithms for these problems should allow the deletion, insertion, and replacement of symbols (nucleotides or amino acids), and they should be capable of comparing a large number of long sequences. An interesting problem related to both sequence alignment and microarray production (see below) is described in Section 16.4.
TABLE 16.1 Main Bioinformatic Tasks and Some Representative Metaheuristics Applied to Them
| Bioinformatic Task | Metaheuristics |
| Sequence comparison and alignment | EA [13,14] MA [15] ACS [16] PSO [17] |
| DNA fragment assembly | EA [18,19] SA [20] ACS [21] |
| Gene finding and identification | EA [22,23] |
| Gene expression profiling | EA [24] MA [25,26] PSO [27] |
| Structure prediction | EA [28,29] MA [30] SA [31] EDA [32] |
| Phylogenetic trees | EA [33,34] SS [35] MA [36] |
DNA Fragment Assembly The fragment assembly problem consists of building the DNA sequence from several hundreds (or even, thousands) of fragments obtained by biologists in the laboratory. This is an important task in any genome project since the rest of the phases depend on the accuracy of the results of this stage. This problem is described in Section 16.3.
Gene Finding and Identification It is frequently the case in bioinformatics that one wishes to delimit parts of sequences that have a biological meaning. Typical examples are determining the locations of promoters, exons, and introns in RNA. In particular, automatic identification of the genes from the large DNA sequences is an important problem. Recognition of regulatory regions in DNA fragments has become particularly popular because of the increasing number of completely sequenced genomes and mass application of DNA chips.
Gene Expression Profiling This is the process for determining when and where particular genes are expressed. Furthermore, the expression of one gene is often regulated by the expression of another gene. A detailed analysis of all this information will provide an understanding of the internetworking of different genes and their functional roles. Microarray technology is used for that purpose.
Microarray technology allows expression levels of thousands of genes to be measured at the same time. This allows the simultaneous study of tens of thousands of different DNA nucleotide sequences on a single microscopic glass slide. Many important biological results can be obtained by selecting, assembling, analyzing, and interpreting microarray data correctly. Clustering is the most common task and allows us to identify groups of genes that share similar expressions and maybe, similar functions.
Structure Prediction Determining the structure of proteins is very important since there exists a strong relation between structure and function. This is one of the most challenging tasks in bioinformatics. There are four main levels of protein structure:
- The primary structure is its linear sequence of amino acids.
- The secondary structure is the folding of the primary structure via hydrogen bonds.
- The tertiary structure refers to the three-dimensional structure of the protein and is generated by packing the secondary structural elements. Generally, the protein function depends on its tertiary structure.
- The quaternary structure describes the formation of protein complexes composed of more than one chain of amino acids.
Also, the protein docking problem is related to the structure of the protein. This problem, to determine the interaction with other proteins, plays a key role in understanding the protein function.
Phylogenetic Analysis All species undergo a slow transformation process called evolution. Phylogenetic trees are labeled binary trees where leaves represent current species and inner nodes represent hypothesized ancestors. Phylogenetic analysis is used to study evolutionary relationships.
16.3 DNA FRAGMENT ASSEMBLY PROBLEM
In this section we study the behavior of several metaheuristics for the DNA fragment assembly problem. DNA fragment assembly is a problem solved in the early phases of the genome project and is thus very important, since the other steps depend on its accuracy. This is an NP-hard combinatorial optimization problem that is growing in importance and complexity as more research centers become involved in sequencing new genomes.
In the next subsection we present background information about the DNA fragment assembly problem. Later, the details of our approaches are presented and also how to design and implement these methods for the DNA fragment assembly problem. We finish this section by analyzing the results of our experiments.
16.3.1 Description of the Problem
To determine the function of specific genes, scientists have learned to read the sequence of nucleotides comprising a DNA sequence in a process called DNA sequencing. To do that, multiple exact copies of the original DNA sequence are made. Each copy is then cut into short fragments at random positions. These are the first three steps depicted in Figure 16.1, and they take place in the laboratory. After the fragment set is obtained, a traditional assemble approach is followed in this order: overlap, layout, and consensus. To ensure that enough fragments overlap, the reading of fragments continues until a coverage is satisfied. These steps are the last three in Figure 16.1. In what follows, we give a brief description of each of the three phases: overlap, layout, and consensus.
Overlap Phase Finding the overlapping fragments. This phase consists of finding the best or longest match between the suffix of one sequence and the prefix of another. In this step we compare all possible pairs of fragments to determine their similarity. Usually, a dynamic programming algorithm applied to semiglobal alignment is used in this step. The intuition behind finding the pairwise overlap is that fragments with a high overlap are very likely to be next to each other in the target sequence.
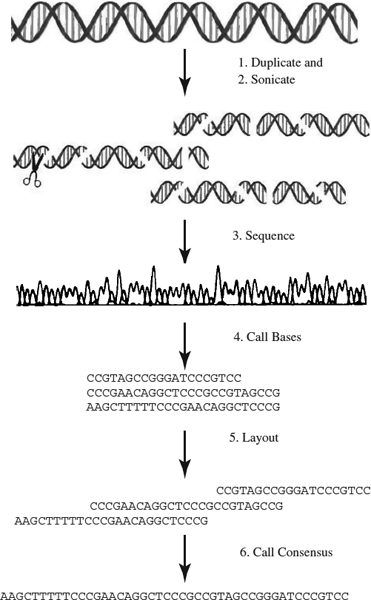
Figure 16.1 Graphical representation of DNA sequencing and assembly.
Layout Phase Finding the order of fragments based on the computed similarity score. This is the most difficult step because it is difficult to tell the true overlap, due to the following challenges:
1. Unknown orientation. After the original sequence is cut into many fragments, the orientation is lost. One does not know which strand should be selected. If one fragment does not have any overlap with another, it is still possible that its reverse complement might have such an overlap.
2. Base call errors. There are three types of base call errors: substitution, insertion, and deletion. They occur due to experimental errors in the electrophoresis procedure (the method used in laboratories to read DNA sequences). Errors affect the detection of fragment overlaps. Hence, the consensus determination requires multiple alignments in high-coverage regions.
3. Incomplete coverage. This occurs when an algorithm is not able to assemble a given set of fragments into a single contig, a sequence in which the overlap between adjacent fragments is greater than or equal to a predefined threshold (cutoff parameter).
4. Repeated regions. Repeats are sequences that appear two or more times in the target DNA. Repeated regions have caused problems in many genome-sequencing projects, and none of the current assembly programs can handle them perfectly.
5. Chimeras and contamination. Chimeras arise when two fragments that are not adjacent or overlapping on the target molecule join into one fragment. Contamination occurs due to incomplete purification of the fragment from the vector DNA.
After the order is determined, the progressive alignment algorithm is applied to combine all the pairwise alignments obtained in the overlap phase.
Consensus Phase Deriving the DNA sequence from the layout. The most common technique used in this phase is to apply the majority rule in building the consensus. To measure the quality of a consensus, we can look at the distribution of the coverage. Coverage at a base position is defined as the number of fragments at that position. It is a measure of the redundancy of the fragment data, and it denotes the number of fragments, on average, in which a given nucleotide in the target DNA is expected to appear. It is computed as the number of bases read from fragments over the length of the target DNA [37]:

where n is the number of fragments. The higher the coverage, the fewer the gaps, and the better the result.
16.3.2 DNA Fragment Assembly Using Metaheuristics
Let us give some details about the most important issues of our implementation and how we have used metaheuristics to solve the DNA fragment assembly problem. First, we address common details such as the solution representation or the fitness function, and then we describe the specific features of each algorithm. The methods used are a genetic algorithm [38], a nontraditional GA called the CHC method [39], scatter search [9], and simulated annealing [4] (for more detail about these algorithms, we refer the reader to the book by Glover and Kochenberger [3].
Common Issues
1. Solution representation. We use the permutation representation with integer number encoding. A permutation of integers represents a sequence of fragment numbers where successive fragments overlap. The solution in this representation requires a list of fragments assigned with a unique integer ID. For example, eight fragments will need eight identifiers: 0, 1, 2, 3, 4, 5, 6, 7. The permutation representation requires special operators to make sure that we always get legal (feasible) solutions. To maintain a legal solution, the two conditions that must be satisfied are: (1) All fragments must be presented in the ordering, and (2) no duplicate fragments are allowed in the ordering.
2. Fitness function. A fitness function is used to evaluate how good a particular solution is. In the DNA fragment assembly problem, the fitness function measures the multiple sequence alignment quality and finds the best scoring alignment. Our fitness function [18] sums the overlap score (w(f, f 1)) for adjacent fragments (f [i] and f [i + 1]) in a given solution. When this fitness function is used, the objective is to maximize such a score. It means that the best individual will have the highest score, since the order proposed by that solution has strong overlap between adjacent fragments.
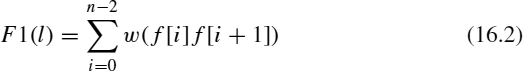
3. Program termination. The program can be terminated in one of two ways. We can specify the maximum number of evaluations to stop the algorithm, or we can stop the algorithm when the solution is no longer improved.
GA Details
1. Population size. We use a fixed-size population of random solutions.
2. Recombination operator. Two or more parents are recombined to produce one or more offspring. The purpose of this operator is to allow partial solutions to evolve in different individuals and then combine them to produce a better solution. It is implemented by running through the population and for each individual, deciding whether it should be selected for crossover using a parameter called crossover rate (Pc). For our experimental runs, we use the order-based crossover (OX). This operator first copies the fragment ID between two random positions in Parent1 into the offspring's corresponding positions. We then copy the rest of the fragments from Parent2 into the offspring in the relative order presented in Parent2. If the fragment ID is already present in the offspring, we skip that fragment. The method preserves the feasibility of every tentative solution in the population.
3. Mutation operator. This operator is used for the modification of single individuals. The reason we need a mutation operator is for the purpose of maintaining diversity in the population. Mutation is implemented by running through the entire population and for each individual, deciding whether or not to select it for mutation, based on a parameter called the mutation rate (Pm). For our experimental runs, we use the swap mutation operator and invert the segment mutation operator. The first operator randomly selects two positions from a permutation and then swaps the two fragment positions. The second one also selects two positions from a permutation and then inverts the order of the fragments in partial permutation defined by the two random positions (i.e., we swap two edges in the equivalent graph). Since these operators do not introduce any duplicate number in the permutation, the solution they produce is always feasible.
4. Selection operator. The purpose of the selection is to weed out bad solutions. It requires a population as a parameter, processes the population using the fitness function, and returns a new population. The level of the selection pressure is very important. If the pressure is too low, convergence becomes very slow. If the pressure is too high, convergence will be premature to a local optimum.
In this chapter we use the ranking selection mechanism [40], in which the GA first sorts individuals based on fitness and then selects the individuals with the best fitness score until the population size specified is reached. Preliminary results favored this method out of a set of other selection techniques analyzed. Note that the population size will grow whenever a new offspring is produced by crossover or mutation operators.
CHC Details
1. Incest prevention. The CHC method has a mechanism of incest prevention to avoid recombination of similar solutions. Typically, the Hamming distance is used as a measure of similarity, but this one is unsuitable for permutations. In the experiments we consider that the distance between two solutions is the total number of edges minus the number of common edges.
2. Recombination. The crossover that we use in our CHC creates a single child by preserving the edges that parents have in common and then randomly assigning the remaining edges to generate a legal permutation.
3. Population restart. Whenever the population converges, the population is partially randomized for a restart. The restart method uses as a template the best individual found so far, creating new individuals by repeatedly swapping edges until a specific fraction of edges differ from those of the template.
SS Details
1. Initial population creation. There exist several ways to get an initial population of good and disperse solutions. In our experiments, the solutions for the population were generated randomly to achieve a certain level of diversity. Then we apply the improvement method to these solutions to get better solutions.
2. Subsets generation and recombination operator. It generates all two-element subsets and then it applies the recombination operator to them. For our experimental runs, we use the order-based crossover that was explained before.
3. Improvement method. We apply a hillclimber procedure to improve the solutions. The hillclimber is a variant of Lin and Kernighan's two-opt [41]. Two positions are selected randomly, and then the subpermutation is inverted by swapping the two edges. Whenever an improvement is found, the solution is updated; the hillclimber continues until it achieves a predetermined number of swap operations.
SA Details
1. Cooling scheme. The cooling schedule controls the values of the temperature parameter. It specifies the initial value and how the temperature is updated at each stage of the algorithm:
In this case we use a decreasing function (Equation 16.3) controlled by the α factor, where α ∈ (0, 1).
2. Markov chain length. The number of the iterations between two consecutive changes of the temperature is given by the parameter Markov chain length, whose name alludes to the fact that the sequence of solutions accepted is a Markov chain (a sequence of states in which each state depends only on the preceding one).
3. Move operator. This operator generates a new neighbor from the current solution. For our experimental runs, we use the edge swap operator. This operator randomly selects two positions from a permutation and inverts the order of the fragments between these two fragment positions.
16.3.3 Experimental Analysis
A target sequence with accession number BX842596 (GI 38524243) was used in this work. It was obtained from the NCBI website (http://www.ncbi.nlm.nih.gov). It is the sequence of a Neurospora crassa (common bread mold) BAC and is 77,292 base pairs long. To test and analyze the performance of our algorithm, we generated two problem instances with GenFrag [42]. GenFrag takes a known DNA sequence and uses it as a parent strand from which to generate fragments randomly according to the criteria (mean fragment length and coverage of parent sequence) supplied by the user. The first problem instance, 842596_4, contains 442 fragments with average fragment length 708 bps and coverage 4. The second problem instance, 842596_7, contains 773 fragments with average fragment length 703 bps and coverage 7. We evaluated the results in terms of the number of contigs assembled.
We use a GA, a CHC, an SS, and an SA to solve this problem. To allow a fair comparison among the results of these heuristics, we have configured them to perform a similar computational effort (the maximum number of evaluations for any algorithm is 512,000). Since the results of these algorithms vary depending on the different parameter settings, we performed a complete analysis previously to study how the parameters affect the performance of the algorithms. A summary of the conditions for our experimentation is given in Table 16.2. We have performed statistical analyses to ensure the significance of the results and to confirm that our conclusions are valid (all the results are different statistically).
Table 16.3 shows all the results and performance with all data instances and algorithms described in this work. The table shows the fitness of the best solution obtained (b), the average fitness found (f), average number of evaluations (e), and average time in seconds (t). We do not show the standard deviation because the fluctuations in the accuracy of different runs are rather small, showing that the algorithms are very robust (as proved by the ANOVA results). The best results are shown in boldface type.
TABLE 16.2 Parameters and Optimum Solutions of the Problem
| Common parameters | |
| Independent runs | 30 |
| Cutoff | 30 |
| Max. evaluations | 51,2000 |
| Genetic algorithms | |
| Population size | 1024 |
| Crossover | OX (0.8) |
| Mutation | Edge swap (0.2) |
| Selection | Ranking |
| CHC | |
| Population size | 50 |
| Crossover | Specific (1.0) |
| Restart | Edge swap (30%) |
| Scatter search | |
| Initial population size | 15 |
| Reference set | 8 (5 + 3) |
| Subset generation | All two-element subsets (28) |
| Crossover | OX (1.0) |
| Improvement | Edge swap (100 iterations) |
| Simulated annealing | |
| Move operator | Edge swap |
| Markov chain length | Total number evaluations/100 |
| Cooling scheme | Proportional (α = 0.99) |
TABLE 16.3 Results for the Two Instances
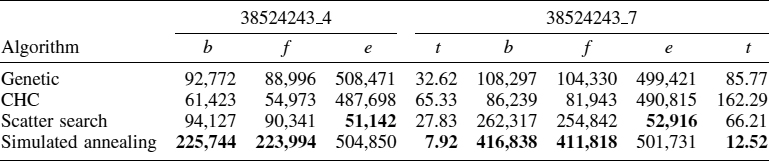
Let us discuss some of the results found in this table. First, for the two instances, it is clear that the SA outperforms the other algorithms from every point of view. In fact, SA obtains better fitness values than do the previous best known solutions [20]. Also, its execution time is the lowest. The reason is that the SA operates on a single solution, while the remaining methods are population based, and in addition, they execute time-consuming operators (especially the crossover operation).
The CHC is the worst algorithm in both execution time and solution quality. Its longer runtime is due to the additional computations needed to detect the converge of the population or to detect incest mating. CHC is not able to solve the DNA fragment assembly problem adequately and perhaps requires a local search (as proposed by Karpenko et al. [16]) to reduce the search space.
The SS obtains better solutions than the GA, and it also achieves these solutions in a shorter runtime. This means that a structured application of operator and explicit reference search points are a good idea for this problem.
The computational effort to solve this problem (considering only the number of evaluations) is similar for all the heuristics with the exception of the SS, because its most time-consuming operation is the improvement method that does not perform complete evaluations of the solutions. This result indicates that all the algorithms examine a similar number of points in the search space, and the difference in the solution quality is due to how they explore the search space. For this problem, trajectory-based methods such as simulated annealing are more effective than population-based methods. Thus, the resulting ranking of algorithms from best to worst is SA, SS, GA, and finally, CHC.
Table 16.4 shows the final number of contigs computed in every case, a contig being a sequence in which the overlap between adjacent fragments is greater than a threshold (cutoff parameter). Hence, the optimum solution has a single contig. This value is used as a high-level criterion to judge the entire quality of the results, since, as we said before, it is difficult to capture the dynamics of the problem in a mathematical function. These values are computed by applying a final step of refinement with a greedy heuristic popular in this application [19]. We have found that in some (extreme) cases it is possible that a solution with a better fitness than other solution generates a larger number of contigs (worse solution). This is the reason for still needing more research to get a more accurate mapping from fitness to contig number. The values of this table, however, confirm again that the SA method outperform the rest clearly, the CHC obtains the worst results, and the SS and GA obtain a similar number of contigs.
TABLE 16.4 Final Best Contigs

16.4 SHORTEST COMMON SUPERSEQUENCE PROBLEM
The shortest common supersequence problem (SCSP) is a classical problem from the realm of string analysis. Roughly speaking, the SCS problem amounts to finding a minimal-length sequence S ∈ Σ* of symbols from a certain alphabet Σ such that every sequence in a certain set L ∈ 2Σ* can be generated from S by removing some symbols of the latter. The resulting combinatorial problem is enormously interesting from the point of view of bioinformatics [43] and bears a close relationship to the sequence alignment and microarray production, among other tasks.
Unfortunately, the SCS problem has been shown to be hard under various formulation and restrictions, resulting not just in a NP-hard problem, but also in a non-FPT problem [44], so practical resolution is probably unaffordable by conventional exact techniques. Therefore, several heuristics and metaheuristics have been defined to tackle it. Before detailing these, let us first describe the SCSP more formally.
16.4.1 Description of the Problem
We write |s| for the length of sequence s(|s1s2 · · · sn| = n) and |Σ| for the cardinality of set Σ. Let ![]() be the empty sequence
be the empty sequence ![]() . We use
. We use ![]() for the total number of occurrences of symbol α in sequence s:
for the total number of occurrences of symbol α in sequence s:
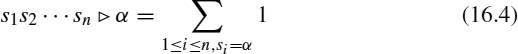
We write αs for the sequence obtained by appending the symbol α in front of sequence s. Deleting the symbol α from the front of sequence s is denoted s|α. We also use the | symbol to delete a symbol from the front of a set of strings: {s1, …, sm}|α = {s1 |α, …, sm|α}.
Sequence s is said to be a supersequence of r (denoted as ![]() ) if
) if ![]() or if s|s1 is a supersequence of r|s1. Plainly,
or if s|s1 is a supersequence of r|s1. Plainly, ![]() implies that r can be embedded in s, meaning that all symbols in r are present in s in exactly the same order, although not necessarily consecutive. We can now state the SCSP as follows: An instance I = (Σ, L) for the SCSP is given by a finite alphabet Σ and a set L of m sequences {s1, …, sm}, si ∈ Σ*. The problem consists of finding a sequence s of minimal length that is a supersequence of each sequence in L (
implies that r can be embedded in s, meaning that all symbols in r are present in s in exactly the same order, although not necessarily consecutive. We can now state the SCSP as follows: An instance I = (Σ, L) for the SCSP is given by a finite alphabet Σ and a set L of m sequences {s1, …, sm}, si ∈ Σ*. The problem consists of finding a sequence s of minimal length that is a supersequence of each sequence in L (![]() , ∀si ∈ L and |s| is minimal). A particularly interesting situation for bioinformatics arises when the sequences represent molecular data (i.e., sequences of nucleotides or amino acids).
, ∀si ∈ L and |s| is minimal). A particularly interesting situation for bioinformatics arises when the sequences represent molecular data (i.e., sequences of nucleotides or amino acids).
16.4.2 Heuristics and Metaheuristics for the SCSP
One of the simplest and most effective algorithms for the SCSP is majority merge (MM). This is a greedy algorithm that constructs a supersequence incrementally by adding the symbol most frequently found at the front of the sequences in L and removing these symbols from the corresponding strings. Ties can be broken randomly, and the process is repeated until all sequences in L are empty. A drawback of this algorithm is its myopic functioning, which makes it incapable of grasping the global structure of strings in L. In particular, MM misses the fact that the strings can have different lengths [45]. This implies that symbols at the front of short strings will have more chances to be removed, since the algorithm still has to scan the longer strings. For this reason, it is less urgent to remove those symbols. In other words, it is better to concentrate first on shortening longer strings. This can be done by assigning a weight to each symbol, depending on the length of the string in whose front it is located. Branke et al. [45] propose using precisely this string length as a weight (i.e., the weight of a symbol is the sum of the length of all strings at whose front it appears). This modified heuristic is termed weighted majority merge (WMM), and its empirical evaluation indicates that it can outperform MM on some problem instances in which there is no structure or where the structure is deceptive [45,46].
The limitations of these simple heuristics have been dealt with via the use of more sophisticated techniques. One of the first metaheuristic approaches to the SCSP is due to Branke et al. [45]. They consider several evolutionary algorithm approaches, in particular a GA that uses WMM as a decoding mechanism. More precisely, the GA evolves weights (metaweights, actually) that are used to further refine the weights computed by the WMM heuristic. The latter algorithm is then used to compute tentative supersequences on the basis of these modified weights. This procedure is similar to the EA used by Cotta and Troya [47] for the multidimensional knapsack problem, in which a greedy heuristic was used to generate solutions, and weights were evolved in order to modify the value of objects. A related approach was presented by Michel and Middendorf [48] based on ACO. Pheromone values take the role of weights, and ants take probabilistic decisions based on these weights. An interesting feature of this ACO approach is the fact that pheromone update is not done on an individual basis (i.e., just on the symbols being picked each time), but globally (i.e., whenever a symbol is picked, the pheromone of previous symbols that allowed that symbol to be picked is also increased).
More recent approaches to the SCSP are based on EAs [46]. These EAs range from simple direct approaches based on penalty functions, to more complex approaches based on repairing mechanisms or indirect encodings:
1. Simple EA. Solutions are represented as a sequence of symbols corresponding to a tentative supersequence. Fitness is computed as the length of the supersequence plus a penalty term in case a solution does not contain a valid supersequence. This penalty term is precisely the length of the solution provided by the MM heuristic for the remaining sequences:
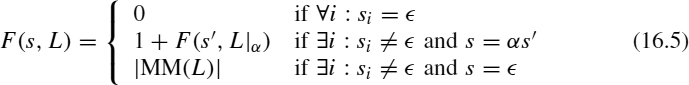
Standard recombination and mutation operators can be used in this EA.
2. EA + repairing. Based on the simple EA, a repair function ρ is used to complete the supersequence with the symbols indicated by the MM heuristic:

Notice that this function not only turns infeasible solutions into feasible ones, but also improves feasible solutions by removing unproductive steps.
3. Indirect EAs. In the line of the GRASP-like decoding mechanism defined by Cotta and Fernández [49], the EA evolves sequences of integers that denote the rank of the symbol that has to be picked at each step, on the basis of the ranking provided by a simple heuristic such as MM or WMM.
The experimental results [46] indicate that the EA with repairing is better than the other two approaches for small alphabet sizes (e.g., in the case of nucleotide sequences). For larger alphabets, the EA with indirect encoding is slightly better. Further improvements are obtained from the inclusion of a local search technique within the EA, thus resulting in a MA [15]. Local search is done by removing symbols from the supersequence and retaining the deletion if the resulting solution is valid. This local search procedure is costly, so it has to be used with caution. To be precise, it is shown that partial Lamarckism rates (around 1 or 5%) provide the best trade-off between computational cost and quality of results.
A completely different metaheuristic, beam search (BS), has also been proposed for the SCSP [50]. This technique approaches the incremental construction of k (a parameter) solutions via a pseudo-population-based greedy mechanism inspired in branch and bound and the breadth-first traversal of search trees. This is a fully deterministic approach (save for tie breaking) that is shown to provide moderately good solutions to the problem. Several improvements to this basic technique have been proposed. On the one hand, the hybridization of BS and MAs has also been attempted [50]. On the other hand, a probabilistic version of BS (PBS) has been described [51]. Both approaches have been shown to be superior to the basic technique and are very competitive in general. PBS is remarkable for its success in small sequences, but it starts to suffer scalability problems when the length of sequences increases.
16.4.3 Experimental Analysis
An experimental comparison has been made between the most competitive algorithms described before: namely, the MA-BS hybrid [50] and both the simple heuristics MM and WMM. The instances considered for the experimentation comprise both DNA sequences (|Σ| = 4) and protein sequences (|Σ| = 20). In the first case we have taken two DNA sequences of the SARS coronavirus from a genomic database (http://gel.ym.edu.tw/sars/genomes.html); these sequences are 158 and 1269 nucleotides long. As to the protein sequences, we have considered three of them, extracted from Swiss-Prot (http://www.expasy.org/sprot/):
- Oxytocin. Quite important in pregnant women, this protein causes contraction of the smooth muscle of the uterus and the mammary gland. The sequence is 125 amino acids long.
- p53. This protein is involved in the cell cycle and acts as tumor suppressor in many tumor types; the sequence is 393 amino acids long.
- Estrogen. Involved in the regulation of eukaryotic gene expression, this protein affects cellular proliferation and differentiation; the sequence is 595 amino acids long.
In all cases, problem instances are constructed by generating strings from the target sequence by removing symbols from the latter with probability p. In our experiments, problem instances comprise 10 strings, and p = {5%, 10%, 20%}, thus representing increasingly difficult optimization problems.
All algorithms are run for 600 seconds on a Pentium-4 2.4-GHz 512-Mbyte computer. The particular parameters of the MA component are population size = 100, pX = 0.9, pM = 1/L, pLS = 0.01, uniform crossover, tournament selection, and steady-state replacement. As to the BS component, it considers k = 10,000. The results (averaged for 20 runs) are shown in Figures 16.2 and 16.3.
As can be seen, the conventional greedy heuristics cannot compete with the hybrid approach. Notice that the results of the latter are very close to the putative optimal (the original sequence to be reconstructed). Actually, it can be seen that the hybrid MA-BS algorithm manages to find this original solution in all runs. As reported by Blum et al. [51], the PBS algorithm is also capable of performing satisfactorily for most of these problem instances but is affected by the increased dimensionality of the problem in the SARS 1269 instance, for high values of p. MA-BS is somewhat more robust in this sense.
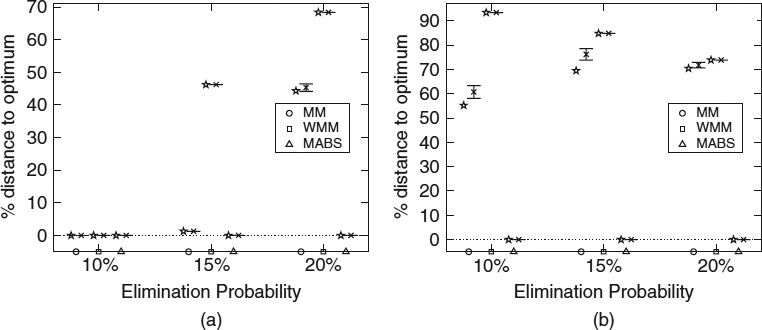
Figure 16.2 Results of MM, WMM, and MA-BS for the (a) SARS158 and (b) SARS1269 instances. Each group of bars indicates the results of the corresponding algorithm (MM, WMM, and MA-BS) for three different values of p (i.e., 5%, 10%, 20%). The star indicates the best solution found by the corresponding algorithm, the cross marks the mean, and the bars indicate the standard deviation.

Figure 16.3 Results of MM, WMM, and MA-BS for the (a) P53 and (b) estrogen instances; all techniques manage to find the optimal solution in most runs for the oxytocin instance. The exceptions are MM and WMM for the 20% case.
16.5 CONCLUSIONS
The increase in biological data and the need to analyze and interpret them have opened several important research lines in computation science since the application of computational approaches allows us to facilitate an understanding of various biological process. In this chapter we have shown how these problems can be solved with metaheuristics. In particular, we have tackled two difficult problems: the DNA fragment assembly problem and the shortest common super-sequence problem.
Both the DNA FAP and the SCSP are very complex problems in computational biology. We tackled the first problem with four sequential heuristics: three population based (GA, SS, and CHC), and one trajectory based (SA). We have observed that the latter outperforms the rest of the methods (which implies more research in trajectory-based methods in the future). It obtains much better solutions than the others do, and it runs faster. This local search algorithm also outperformed the best known fitness values in the literature. As to the second, we have shown that the best approach described in the literature is the hybrid of memetic algorithms and beam search defined by Gallardo et al. [50]. This hybrid method vastly outperforms conventional greedy heuristics on both DNA sequences and protein sequences.
Acknowledgments
The authors are partially supported by the Ministry of Science and Technology and FEDER under contract TIN2005-08818-C04-01 (the OPLINK project). The second and third authors are also supported under contract TIN-2007-67134.
REFERENCES
1. J. Cohen. Bioinformatics: an introduction to computer scientists. ACM Computing Surveys, 36:122–158, 2004.
2. C. Blum and A. Roli. Metaheuristics in combinatorial optimization: overview and conceptual comparison. ACM Computing Surveys, 35(3):268–308, 2003.
3. F. Glover and A. G. Kochenberger. Handbook of Metaheuristics. International Series in Operations Research and Management Science. Springer-Verlag, New York, Jan. 2003.
4. S. Kirkpatrick, C. D. Gelatt, and M. P. Vecchi. Optimization by simulated annealing. Science, 220(4598):671–680, 1983.
5. F. Glover. Tabu search: I. ORSA, Journal of Computing, 1:190–206, 1989.
6. N. Mladenovic and P. Hansen. Variable neighborhood search. Computers and Operations Research, 24:1097–1100, 1997.
7. T. Bäck, D. B. Fogel, and Z. Michalewicz, eds. Handbook of Evolutionary Computation. Oxford University Press, New York, 1997.
8. M. Dorigo and T. Stützle. Ant Colony Optimization. MIT Press, Cambridge, MA, 2004.
9. F. Glover, M. Laguna, and R. Martí. Fundamentals of scatter search and path relinking. Control and Cybernetics, 39(3):653–684, 2000.
10. P. Moscato and C. Cotta. A gentle introduction to memetic algorithms. In F. Glover and G. Kochenberger, eds., Handbook of Metaheuristics. Kluwer Academic, Norwell, MA, 2003, pp. 105–144.
11. P. Moscato, C. Cotta, and A. Mendes. Memetic algorithms. In G. C. Onwubolu and B. V. Babu, eds., New Optimization Techniques in Engineering. Springer-Verlag, New York, 2004, pp. 53–85.
12. P. Moscato and C. Cotta. Memetic algorithms. In T. González, ed., Handbook of Approximation Algorithms and Metaheuristics. Taylor & Francis, London, 2007.
13. C. Notredame and D. G. Higgins. SAGA: sequence alignment by genetic algorithm. Nucleic Acids Research, 24:1515–1524, 1996.
14. C. Zhang and A. K. Wong. A genetic algorithm for multiple molecular sequence alignment. Bioinformatics, 13(6):565–581, 1997.
15. C. Cotta. Memetic algorithms with partial Lamarckism for the shortest common super-sequence problem. In J. Mira and J. R. Álvarez, eds., Artificial Intelligence and Knowledge Engineering Applications: A Bioinspired Approach, vol. 3562 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2005, pp. 84–91.
16. O. Karpenko, J. Shi, and Y. Dai. Prediction of MHC class II binders using the ant colony search strategy. Artificial Intelligence in Medicine, 35(1–2):147–156, 2005.
17. T. K. Rasmussen and T. Krink. Improved hidden Markov model training for multiple sequence alignment by a particle swarm optimization–evolutionary algorithm hybrid. Biosystems, 72(1–2):5–17, 2003.
18. R. Parsons, S. Forrest, and C. Burks. Genetic algorithms, operators, and DNA fragment assembly. Machine Learning, 21:11–33, 1995.
19. L. Li and S. Khuri. A comparison of DNA fragment assembly algorithms. In Proceedings of the International Conference on Mathematics and Engineering Techniques in Medicine and Biological Sciences, 2004, pp. 329–335.
20. E. Alba, G. Luque, and S. Khuri. Assembling DNA fragments with parallel algorithms. In B. McKay, ed., Proceedings of the 2005 Congress on Evolutionary Computation, Edinburgh, UK, 2005, pp. 57–65.
21. P. Meksangsouy and N. Chaiyaratana. DNA fragment assembly using an ant colony system algorithm. In Proceedings of the 2003 Congress on Evolutionary Computation, vol. 3. IEEE Press, New York 2003, pp. 1756–1763.
22. A. Kel, A. Ptitsyn, V. Babenko, S. Meier-Ewert, and H. Lehrach. A genetic algorithm for designing gene family-specific oligonucleotide sets used for hybridization: the g protein-coupled receptor protein superfamily. Bioinformatics, 14:259–270, 1998.
23. V. G. Levitsky and A. V. Katokhin. Recognition of eukaryotic promoters using a genetic algorithm based on iterative discriminant analysis. In Silico Biology, 3(1):81–87, 2003.
24. K. Deb, S. Agarwal, A. Pratap, and T. Meyarivan. A fast elitist non-dominated sorting genetic algorithm for multi-objective optimization: NSGA-II. In Proceedings of the Parallel Problem Solving from Nature VI, 2000, pp. 849–858.
25. C. Cotta, A. Mendes, V. Garcia, P. Franca, and P. Moscato. Applying memetic algorithms to the analysis of microarray data. In G. Raidl et al., eds., Applications of Evolutionary Computing, vol. 2611 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2003, pp. 22–32.
26. A. Mendes, C. Cotta, V. Garcia, P. França, and P. Moscato. Gene ordering in microarray data using parallel memetic algorithms. In T. Skie and C.-S. Yang, eds., Proceedings of the 2005 International Conference on Parallel Processing Workshops, Oslo, Norway. IEEE Press, New York, 2005, pp. 604–611.
27. X. Xiao, E. R. Dow, R. Eberhart, Z. B. Miled, and R. J. Oppelt. Gene clustering using self-organizing maps and particle swarm optimization. In Proceedings of the 6th IEEE International Workshop on High Performance Computational Biology, Nice, France, Apr. 2003.
28. C. Notredame, L. Holm, and D. G. Higgins. COFFEE: an objective function for multiple sequence alignments. Bioinformatics, 14(5):407–422, 1998.
29. G. Fogel and D. Corne. Evolutionary Computation in Bioinformatics. Morgan Kaufmann, San Francisco, CA, 2002.
30. C. Cotta. Protein structure prediction using evolutionary algorithms hybridized with backtracking. In J. Mira and J. R. Álvarez, eds., Artificial Neural Nets Problem Solving Methods, vol. 2687 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2003, pp. 321–328.
31. J. J. Gray, S. Moughon, C. Wang, O. Schueler-Furman, B. Kuhlman, C. A. Rohl, and D. Baker. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. Journal of Molecular Biology, 331(1):281–299, 2003.
32. R. Santana, P. Larrañaga, and J. A. Lozano. Protein folding in 2-dimensional lattices with estimation of distribution algorithms. In J. M Barreiro, F. Martín-Sánchez, V. Maojo, and F. Sanz, eds., Proceedings of the 5th Biological and Medical Data Analysis International Symposium, vol. 3337 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2004, pp. 388–398.
33. P. O. Lewis. A genetic algorithm for maximum likelihood phylogeny inference using nucleotide sequence data. Molecular Biology and Evolution, 15:277–283, 1998.
34. C. Cotta and P. Moscato. Inferring phylogenetic trees using evolutionary algorithms. In J. J. Merelo et al., eds., Parallel Problem Solving from Nature VII, vol. 2439 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2002, pp. 720–729.
35. C. Cotta. Scatter search with path relinking for phylogenetic inference. European Journal of Operational Research, 169(2):520–532, 2006.
36. J. E. Gallardo, C. Cotta, and A. J. Fernández. Reconstructing phylogenies with memetic algorithms and branch and bound. In S. Bandyopadhyay, U. Maulik, and J. T. L. Wang, eds., Analysis of Biological Data: A Soft Computing Approach. World Scientific, Hackensack, NJ, 2007, pp. 59–84.
37. J. Setubal and J. Meidanis. Fragment assembly of DNA. In Introduction to Computational Molecular Biology. University of Campinas, Brazil, 1997, pp. 105–139.
38. J. H. Holland. Adaptation in Natural and Artificial Systems. University of Michigan Press, Ann Arbor, MI, 1975.
39. L. J. Eshelman. The CHC adaptive search algorithm: how to have safe search when engaging in nontraditional genetic recombination. In Foundations of Genetic Algorithms, Vol. 1. Morgan Kaufmann, San Francisco, CA, 1991, pp. 265–283.
40. D. Whitely. The GENITOR algorithm and selection pressure: why rank-based allocation of reproductive trials is best. In J. D. Schaffer, ed., Proceedings of the 3rd International Conference on Genetic Algorithms. Morgan Kaufmann, San Francisco, CA, 1989, pp. 116–121.
41. S. Lin and B. W. Kernighan. An effective heuristic algorithm for TSP. Operations Research, 21:498–516, 1973.
42. M. L. Engle and C. Burks. Artificially generated data sets for testing DNA fragment assembly algorithms. Genomics, 16, 1993.
43. M. T. Hallet. An integrated complexity analysis of problems from computational biology. Ph.D. thesis, University of Victoria, 1996.
44. R. Downey and M. Fellows. Parameterized Complexity. Springer-Verlag, New York, 1998.
45. J. Branke, M. Middendorf, and F. Schneider. Improved heuristics and a genetic algorithm for finding short supersequences. OR-Spektrum, 20:39–45, 1998.
46. C. Cotta. A comparison of evolutionary approaches to the shortest common supersequence problem. In J. Cabestany, A. Prieto, and D. F. Sandoval, eds., Computational Intelligence and Bioinspired Systems, vol. 3512 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2005, pp. 50–58.
47. C. Cotta and J. M. Troya. A hybrid genetic algorithm for the 0–1 multiple knapsack problem. In G. D. Smith, N. C. Steele, and R. F. Albrecht, eds., Artificial Neural Nets and Genetic Algorithms, Vol. 3. Springer-Verlag, New York, 1998, pp. 251–255.
48. R. Michel and M. Middendorf. An ant system for the shortest common supersequence problem. In D. Corne, M. Dorigo, and F. Glover, eds., New Ideas in Optimization. McGraw-Hill, London, 1999, pp. 51–61.
49. C. Cotta and A. Fernández. A hybrid GRASP-evolutionary algorithm approach to Golomb ruler search. In X. Yao et al., eds., Parallel Problem Solving from Nature VIII, vol. 3242 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2004, pp. 481–490.
50. J. E. Gallardo, C. Cotta, and A. J. Fernández. Hybridization of memetic algorithms with branch-and-bound techniques. IEEE Transactions on Systems, Man, and Cybernetics, Part B, 37(1):77–83, 2007.
51. C. Blum, C. Cotta, A. J. Fernández, and J. E. Gallardo. A probabilistic beam search approach to the shortest common supersequence problem. In C. Cotta and J. van Hemert, eds., Evolutionary Computation in Combinatorial Optimization, vol. 4446 of Lecture Notes in Computer Science. Springer-Verlag, New York, 2007, pp. 36–47.
Optimization Techniques for Solving Complex Problems, Edited by Enrique Alba, Christian Blum, Pedro Isasi, Coromoto León, and Juan Antonio Gómez
Copyright © 2009 John Wiley & Sons, Inc.